de novo转录组 流程_AI-De Novo分子设计
de novo转录组 流程
大海捞针 (Needle in a Haystack)
On average, it takes ten years and costs $2.6 billion dollars to take a drug from the point of understanding the root cause of a disease to its availability in the marketplace. A large portion of this time and effort/cost is because we are literally looking for a needle in a haystack. We are looking for the one molecule that can turn off a disease at the molecular level in a solution space of between 10³⁰ to a google (yes, 10¹⁰⁰) synthetically feasible molecules. The chemical solution space is too vast to be efficiently screened for the particular molecule of interest. Pharmaceutical compound repositories contain only a fraction of the synthetically feasible molecules for research in a wet lab.
从了解疾病的根本原因到在市场上可获得这种药物,平均花费十年时间,花费26亿美元。 这种时间和精力/成本的很大一部分是因为我们实际上是在大海捞针中寻找针头。 我们正在寻找一种可以在分子水平上以10³3到谷歌(是的10´1)合成可行分子的溶液空间关闭疾病的分子。 化学溶液空间太大,无法有效筛选特定的目标分子。 药物化合物存储库仅包含可在湿实验室中进行研究的合成可行分子的一部分。
Computational de novo drug design can be used as a tool to explore this vast chemical space and synthesize new, never before designed, molecules. Computational drug design can greatly reduce the time spent in the discovery phase; thereby enabling both faster time to market and lower medicine costs.
从头计算药物设计可以用作探索这一巨大化学空间并合成从未设计过的新分子的工具。 计算药物设计可以大大减少在发现阶段花费的时间; 从而缩短上市时间并降低药品成本。
目标 (The Target)
The majority of COVID-19 media images have been of the coronavirus with its protruding spikes. These spikes are the S-proteins that bind to human cells and allow the virus to infect them.
大多数COVID-19媒体图像都是冠状病毒,带有突出的尖峰。 这些尖峰是与人类细胞结合并允许病毒感染它们的S蛋白。

When the virus fuses with a cell, viral genetic material is released into the cell in the form of viral RNA. The viral RNA highjacks our own cells to create the viral proteins needed to make additional copies of itself. Those copies are then released from the cell and used to infect other cells. There is an important protein involved in this process, the main protease. The main protease is responsible for creating the functional proteins needed to assemble other copies of the virus.
当病毒与细胞融合时,病毒遗传物质以病毒RNA的形式释放到细胞中。 病毒RNA劫持了我们自己的细胞,以产生制造自身额外拷贝所需的病毒蛋白。 这些拷贝然后从细胞中释放出来并用于感染其他细胞。 有一个重要的蛋白质参与这一过程,即主要的蛋白酶。 主要蛋白酶负责产生装配病毒其他拷贝的功能蛋白。
If we can find a drug that binds with the main protease and stops it from creating new viral proteins, viral replication can be slowed or stopped. In this article, we explore use of deep learning AI for generating new molecules (aka ligands) that bind to the COVID-19 main protease.
如果我们发现一种与主要蛋白酶结合并阻止其产生新病毒蛋白的药物,则可以减慢或停止病毒复制。 在本文中,我们探索了使用深度学习AI来生成与COVID-19主要蛋白酶结合的新分子(也称为配体)的方法。

解决方案概述 (Solution Overview)
The approach is to create an AI deep learning neural network that will learn how to create ligand molecules. Then a generative model is built from the trained neural network to design new synthetic molecules. AutoDock Vina is used to perform virtual screening of the new molecules to assess their effectiveness for binding to the COVID-19 main protease.
该方法是创建一个AI深度学习神经网络,该网络将学习如何创建配体分子。 然后,从受过训练的神经网络建立一个生成模型,以设计新的合成分子。 AutoDock Vina用于对新分子进行虚拟筛选,以评估其与COVID-19主蛋白酶结合的有效性。
The generated molecules with the best binding scores are then used as the transfer learning dataset to further refine the neural network model toward creating more effective inhibitors of the COVID-19 main protease. Using the updated model, a second generation of molecules is created and screened for their binding assessment.
然后将生成的具有最佳结合分数的分子用作转移学习数据集,以进一步完善神经网络模型,以创建更有效的COVID-19主蛋白酶抑制剂。 使用更新的模型,可以创建第二代分子,并对其结合评估进行筛选。
The high-level workflow:
高级工作流程:
- Obtain and prepare the molecule training data获取并准备分子训练数据
- Create a neural network model and train it to make molecules创建一个神经网络模型并对其进行训练以制造分子
- Develop a generative AI model and create new molecules开发生成型AI模型并创建新分子
- Virtually screen the new molecules against the COVID-19 main protease虚拟筛选针对COVID-19主蛋白酶的新分子
- Use transfer learning to train the neural network on a more specific task使用转移学习在更具体的任务上训练神经网络
- Generate new AI molecules using the generative model使用生成模型生成新的AI分子
- Virtually screen the new molecules against the COVID-19 main protease虚拟筛选针对COVID-19主蛋白酶的新分子
分子数据 (Molecule Data)
The molecule data used to train our neural network will be in the Simplified Molecular Input Line Entry System or ‘SMILES’ format. This is a specification for describing the structure of chemical entities using a combination of characters, numbers and special characters. It’s all about the SMILES :)
用于训练我们的神经网络的分子数据将采用简化分子输入线输入系统或“ SMILES”格式。 这是一个使用字符,数字和特殊字符的组合来描述化学实体结构的规范。 这一切都是关于微笑:)
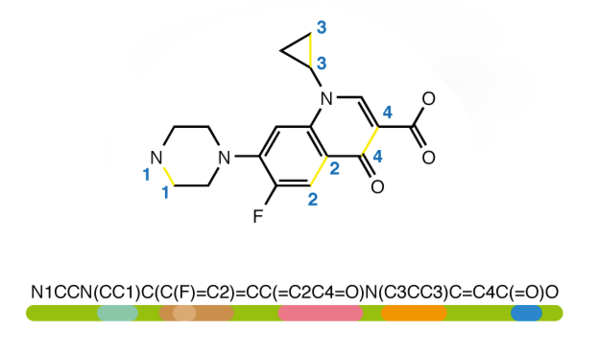
439,098 molecules were obtained from a combination of the Moses and ChEMBL datasets. The SMILES data was cleansed of any salts, charges and stereochemical information using RDKIT — an open-source Cheminformatics software package with a Python API. All of the code and data for this article can be found in my GitHub repo.
从Moses和ChEMBL数据集的组合中获得了439,098个分子。 使用RDKIT (带有Python API的开源Cheminformatics软件包)可以清除SMILES数据中的盐,电荷和立体化学信息。 可以在我的GitHub存储库中找到本文的所有代码和数据。
The SMILES character strings must be vectorized into one-hot encoded arrays of 1’s and 0’s in order to be digitally processed through the neural network. To prep for this, a character set is built from all characters in the SMILES strings. The character set is then used to create two Python dictionaries that facilitate the translation back and forth between characters and numbers.
为了通过神经网络进行数字处理,必须将SMILES字符串向量化为1和0的一键编码数组。 为此,将根据SMILES字符串中的所有字符来构建字符集。 然后使用字符集创建两个Python字典,以方便在字符和数字之间来回转换。
Since the end goal is to create new molecules, we need to include markers in the training data SMILES strings that allow the neural network to learn when it’s starting the creation of a new molecule and when it has reached the end of generating that molecule. Therefore, we manually add molecule start (!) and stop (E) characters to our character set.
由于最终目标是创建新分子,因此我们需要在训练数据SMILES字符串中包含标记,以使神经网络能够了解何时开始创建新分子以及何时到达生成该分子的终点。 因此,我们将分子的开始(!)和停止(E)字符手动添加到我们的字符集中。
AI神经网络 (AI Neural Network)
The neural network will be a sequence-to-sequence model that learns how to reproduce molecules one character at a time. The model will utilize an encoder / decoder architecture. The encoder model constricts the dimensionality of the processing space to create a compressed representation of the input feature characteristics. The decoder model then learns to create the output from that compressed representation.
神经网络将是一个序列到序列的模型,该模型学习如何一次复制一个字符的分子。 该模型将利用编码器/解码器架构。 编码器模型限制了处理空间的维度,以创建输入特征的压缩表示。 然后,解码器模型学习从该压缩表示形式创建输出。
Since the molecule data is a sequence of characters, the AI model is created using Long Short Term Memory (LSTM) cells. LSTM’s are a specific type of Recurrent Neural Network (RNN) cell that have been successfully used to solve a number of sequence related AI tasks; such as natural language processing, voice and text translation, music composition and more recently, the field of molecular informatics.
由于分子数据是字符序列,因此使用长短期记忆(LSTM)单元创建AI模型。 LSTM是一种特定类型的递归神经网络(RNN)单元,已成功用于解决许多与序列相关的AI任务; 例如自然语言处理,语音和文本翻译,音乐创作,以及最近的分子信息学领域。
The neural network models were created using Python 3.7, TensorFlow 2.2 and the Keras high-level programming framework. The models were trained using Google’s Colab with a Tesla P100 GPU. While there is a free version of Google Colab, the paid version enables longer run times and access to significantly faster GPU’s; well worth the money.
使用Python 3.7,TensorFlow 2.2和Keras高级编程框架创建了神经网络模型。 使用Google的Colab和Tesla P100 GPU对模型进行了训练。 虽然有免费的Google Colab版本,但付费版本可以延长运行时间,并可以显着提高GPU的访问速度; 非常值得的钱。
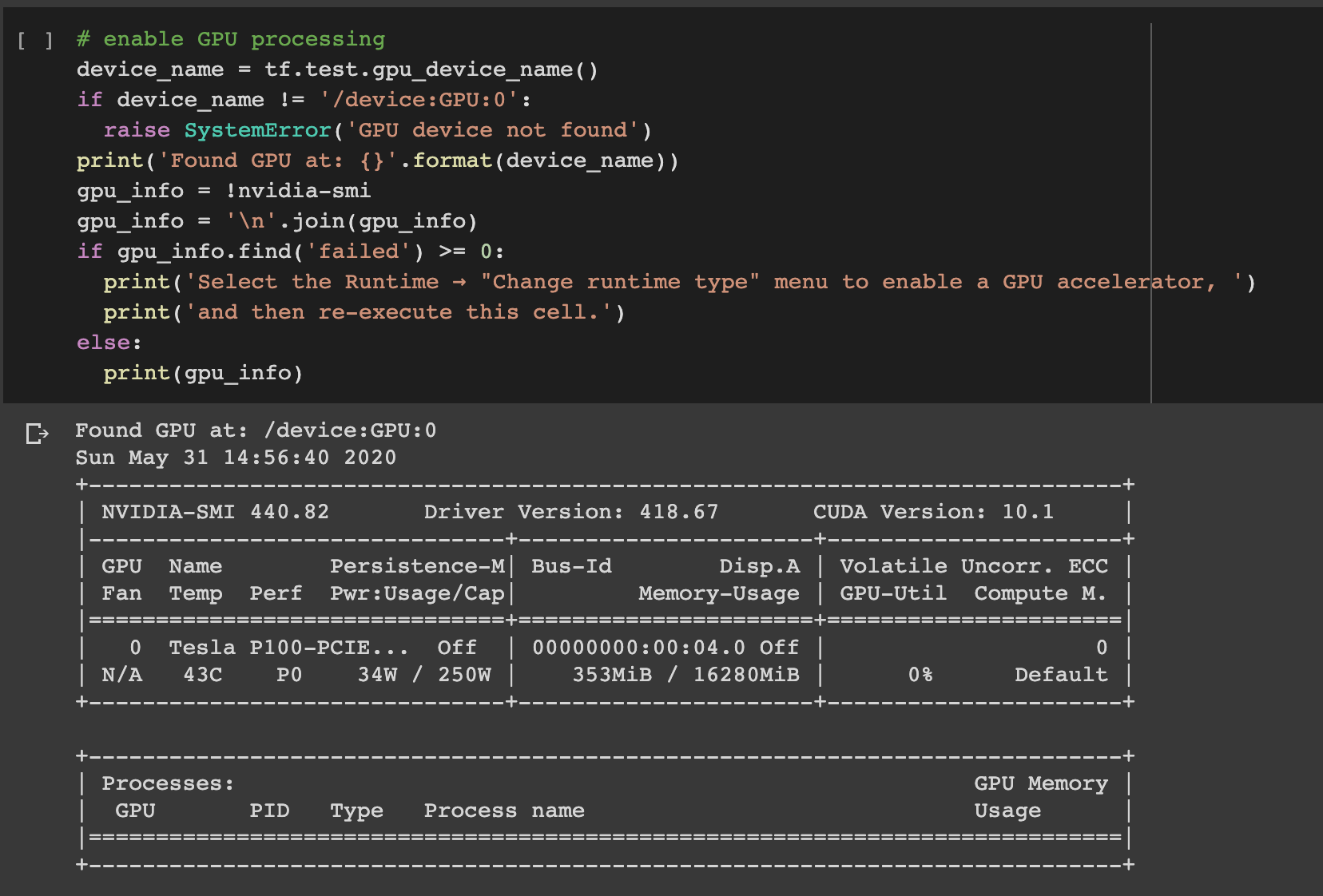
A key first step within Google Colab, is to set your Jupyter notebook settings: set hardware acceleration to GPU and the runtime shape to High-RAM. You can use the code below to enable your GPU and mount your Google Drive.
Google Colab的关键第一步是设置Jupyter笔记本设置:将硬件加速设置为GPU,将运行时形状设置为High-RAM。 您可以使用以下代码启用GPU并挂载Google云端硬盘。
# mount Google Drivefrom google.colab import drivedrive.mount('/drive')The SMILES input data needs to be transformed into a format suitable for supervised learning. A function is used to add the beginning (!) and ending character (E) markers, add padding for constant sequence length and transform each SMILES sequence into a sequence of one-hot encoded vectors.
SMILES输入数据需要转换为适合监督学习的格式。 使用一个函数来添加开始(!)和结束字符(E)标记,添加用于恒定序列长度的填充,并将每个SMILES序列转换为一个一键编码的矢量序列。

LSTM neural networks expect your input data to be a 3D tensor of shape (number of samples, number of time steps, number of features). Here, we have chosen a time step value of 100. This means that:
LSTM神经网络希望您的输入数据是形状的3D张量(样本数,时间步数,特征数)。 在这里,我们选择的时间步长值为100。这意味着:
- The maximum input and output length of our molecule sequences are 100 characters in length我们的分子序列的最大输入和输出长度为100个字符
- During training, all input data will be 100 characters long with the ‘E’ character acting as padding from the end of the SMILES molecule to 100在训练期间,所有输入数据的长度将为100个字符,并且从SMILES分子末端到100的'E'字符用作填充
The number of features equates to the length of our character set, 45.
功能的数量等于字符集的长度45。

Now we define our neural network model.
现在我们定义我们的神经网络模型。

Based on the input tensor, the encoder model obtains the hidden and cell states from a single LSTM layer. Note that we discard the outputs of the LSTM layer, only capturing the state. This state will be used to provide the initial context for the decoder model. The states are then concatenated and used as input to a dense layer that outputs a compressed representation of our initial input.
基于输入张量,编码器模型从单个LSTM层获得隐藏状态和单元状态。 请注意,我们丢弃LSTM层的输出,仅捕获状态。 该状态将用于为解码器模型提供初始上下文。 然后将状态连接起来并用作密集层的输入,该密集层输出我们初始输入的压缩表示。
The decoder model extracts the LSTM hidden and cell states from the compressed data. Those states are then used as input conditioning to an LSTM processing layer. Finally, we use a dense softmax layer to generate the model output.
解码器模型从压缩数据中提取LSTM隐藏和单元状态。 然后将这些状态用作LSTM处理层的输入条件。 最后,我们使用密集的softmax层来生成模型输出。
An architecture summary of the neural network model is provided below.
下面提供了神经网络模型的体系结构摘要。

The model was compiled using the Adam optimizer and categorical crossentropy for our loss function. Due to the quantity of training data, Python generators were used to batch feed the training data to the model.
使用亚当优化器和分类交叉熵为损失函数编译模型。 由于训练数据量大,因此使用Python生成器将训练数据批量输入模型。
The model was trained for 225 epochs, yielding a 99.86% training accuracy and 99.63% validation accuracy. The model achieved 99.66% accuracy on the test dataset.
对模型进行了225个时期的训练,产生了99.86%的训练准确度和99.63%的验证准确度。 该模型在测试数据集上达到了99.66%的准确性。

AI模型分解 (AI Model Decomposition)
The baseline model is now broken up into constituent parts that will be used to build a generative model. If you recall, the original model architecture consisted of two connected neural network models — an encoder and a decoder. Now that the two-part model has been trained in a coordinated manner, we break the respective models apart and add a third, intermediary model.
现在,将基线模型分解为组成部分,这些部分将用于构建生成模型。 您还记得吗,原始模型架构由两个连接的神经网络模型组成-编码器和解码器。 既然已经对两部分模型进行了协调训练,我们将各个模型分开,并添加第三个中间模型。
First, we create a standalone encoder model from our base model.
首先,我们从基本模型创建一个独立的编码器模型。

Next, we create an intermediary model; one that can decode the latent space generated by our encoder model into the LSTM states needed as input to our decoder model. For this model, a new input layer is defined that matches the latent space output by the encoder. The layers of the base model can be used to obtain the LSTM hidden and cell states. This approach allows us to inherit the weights from the previously trained model.
接下来,我们创建一个中介模型; 可以将我们的编码器模型生成的潜在空间解码为LSTM状态,以作为我们的解码器模型的输入。 对于此模型,定义了一个新的输入层,该层与编码器输出的潜在空间匹配。 基本模型的各层可用于获取LSTM隐藏状态和单元状态。 这种方法使我们可以从先前训练的模型继承权重。

Now create the standalone decoder model. While the base model was trained in a stateless batch mode, here, we set the decoder model to be stateful so as to predict one SMILES character at a time. The neural network model layers are defined exactly as before, except with a new input batch_shape (which is required for stateful LSTM model). We then transfer the trained weights from our base model to the standalone decoder model.
现在创建独立的解码器模型。 虽然基本模型是在无状态批处理模式下训练的,但在这里,我们将解码器模型设置为有状态的,以便一次预测一个SMILES字符。 除了使用新的输入batch_shape(有状态LSTM模型所需)外,神经网络模型层的定义与以前完全相同。 然后,我们将训练后的权重从基本模型转移到独立的解码器模型。

Since this model will predict one SMILES character at a time, we use an input layer of shape: samples = 1, time steps =1, features=45.
由于此模型将一次预测一个SMILES字符,因此我们使用形状的输入层:样本= 1,时间步长= 1,特征= 45。
De Novo分子世代 (De Novo Molecule Generation)
Now the fun part — we’ll use our deep learning models to create new, potentially never before designed, molecules!
现在最有趣的部分–我们将使用深度学习模型来创建可能从未设计过的新分子!
For generating new molecules, we create a custom function that allows us to play with the temperature of the Softmax activation function during molecule creation. Decreasing the temperature from 1 to some lower number (e.g. 0.5) makes the model more conservative with its samples. Conversely, a higher temperature (> 1), will provide both more diversity in our samples, but also increase the number of malformed / invalid molecules generated. Conceptually, you can think of this as injecting varying levels of mutation.
为了生成新分子,我们创建了一个自定义函数,该函数允许我们在分子创建过程中使用Softmax激活函数的温度。 将温度从1降低到较低的数字(例如0.5)会使模型对其样本更加保守。 相反,较高的温度(> 1),不仅可以为我们的样品提供更多的多样性,还可以增加生成的畸形/无效分子的数量。 从概念上讲,您可以将其视为注入不同水平的突变。

For more information on this functionality, please check out Andrej Karpathy’s blog (yes, of Tesla AI).
有关此功能的更多信息,请查看Andrej Karpathy的博客 (是,来自Tesla AI)。
To generate new molecules from the encoder generated latent space, we need to compute the LSTM hidden and cell states using the latent_to_states_model and then use them as the input states to the generative model. In inference mode, the generative model will be fed one input character at a time and iteratively sample the next character until the end character is encountered.
为了从编码器生成的潜在空间生成新分子,我们需要使用latent_to_states_model计算LSTM隐藏和单元状态,然后将它们用作生成模型的输入状态。 在推断模式下,将一次向生成模型提供一个输入字符,并迭代采样下一个字符,直到遇到结束字符为止。

To validate the ability to create molecules, we load the test dataset and run it through the encoder model to generate a latent space. We then compare the generated and test set molecules at different sampling temperatures.
为了验证创建分子的能力,我们加载测试数据集并通过编码器模型运行它以生成潜在空间。 然后,我们比较在不同采样温度下生成的和测试集的分子。

The SMILES strings match at both sampling temperatures. Next, we assess the percentage of invalid SMILES molecules created by our generative model.
SMILES琴弦在两个采样温度下均匹配。 接下来,我们评估由我们的生成模型创建的无效SMILES分子的百分比。

As expected, we see an increase in the number of invalid molecules generated at the higher sampling temperature.
如预期的那样,我们看到在较高的采样温度下生成的无效分子的数量有所增加。
Using the ‘sample_smiles’ function, we develop a function to generate molecules around a seed vector in latent space. The function also checks the validity of the generated molecules and only returns valid molecules.
使用“ sample_smiles”函数,我们开发了一种在潜在空间中的种子向量周围生成分子的函数。 该函数还检查生成的分子的有效性,仅返回有效的分子。


Finally, we create a “FOR” loop to generate a batch of SMILES molecules using seed vectors in our latent space.
最后,我们使用潜在空间中的种子向量创建一个“ FOR”循环以生成一批SMILES分子。

This selects a random Softmax activation temperature between 0.75 and 1.25 and attempts to generate up to 50 valid molecules in the vector space around each seed.
这将在0.75和1.25之间选择一个随机的Softmax激活温度,并尝试在每个种子周围的向量空间中生成多达50个有效分子。
创建COVID-19配体 (Create COVID-19 Ligands)
The dataset used for generating our latent space seed vectors is a set of 500 molecules known to be effective inhibitors against the RNA dependent RNA polymerase (RdRp) of the SARS-CoV virus. A RdRp protein is present in all RNA viruses that lack a DNA stage; this includes the Zika virus, Ebola virus and coronaviruses. The dataset also included a handful of HIV inhibitor ligands.
用于生成我们的潜在空间种子载体的数据集是一组500个分子,已知是有效抵抗SARS-CoV病毒的RNA依赖性RNA聚合酶(RdRp)的抑制剂。 在缺少DNA阶段的所有RNA病毒中都存在RdRp蛋白; 这包括寨卡病毒,埃博拉病毒和冠状病毒。 该数据集还包括少数HIV抑制剂配体。
The molecule generation loop was executed three times generating 4,927 new SMILES molecules that were saved for virtual screening.
执行了三次分子生成循环,生成了4,927个新的SMILES分子,这些分子已保存用于虚拟筛选。
对接分析 (Docking Analysis)
To prepare the molecules for the virtual screening process, you will need to install both Open Babel and PyMOL. Open Babel is an open source chemistry toolbox that supports both a graphical and command line interface. PyMOL is an open source molecular visualization system.
要准备用于虚拟筛选过程的分子,您将需要同时安装Open Babel和PyMOL。 Open Babel是一个开放源化学工具箱,它同时支持图形和命令行界面。 PyMOL是一个开源分子可视化系统。
AutoDock Vina, from the Scripps Research Institute, is used to perform the virtual docking analysis between the target protein (COVID-19 main protease) and our ligands (the AI generated molecules). A set of Python scripts developed by Sari Sabban (AutoDock.py) and a few created by me will assist with the virtual screening process.
来自斯克里普斯研究所(Scripps Research Institute)的AutoDock Vina用于在目标蛋白(COVID-19主要蛋白酶)和我们的配体(由AI产生的分子)之间进行虚拟对接分析。 由Sari Sabban (AutoDock.py)开发的一组Python脚本以及由我创建的一些Python脚本将有助于虚拟筛选过程。
准备目标 (Prepare Target)
The first step of virtual screening is to download and prepare the target protein for the docking analysis. The COVID-19 main protease can be downloaded from the RCSB Protein Data Bank. Search for 6LU7 and download it in the PDB (Protein Data Bank) format.
虚拟筛选的第一步是下载并准备目标蛋白以进行对接分析。 可以从RCSB蛋白质数据库中下载COVID-19主蛋白酶。 搜索6LU7并以PDB(蛋白质数据库)格式下载。
Now use the command below to extract just the atoms out of the protein structure.
现在,使用下面的命令从蛋白质结构中仅提取原子。
grep ATOM 6lu7.pdb > temp.pdb The file needs to be converted into the .pdbqt format for use with AutoDock Vina; .pdbqt files are a protein data bank files with a charge applied to every atom in the structure. Use the below command to execute the conversion.
该文件需要转换为.pdbqt格式才能与AutoDock Vina一起使用; .pdbqt文件是蛋白质数据库文件,具有对结构中的每个原子施加电荷的功能。 使用以下命令执行转换。
python AutoDock.py -r temp.pdbThis will output a file called receptor.pdbqt; this is the target protein for our docking analysis.
这将输出一个名为receptor.pdbqt;的文件。 这是我们对接分析的目标蛋白。
The next step is to determine your docking analysis search space. This is the search area upon which AutoDock Vina will assess the ability of a ligand to bind with a target protein. Run the following command in a terminal window.
下一步是确定对接分析搜索空间。 这是AutoDock Vina将在其上评估配体与靶蛋白结合的能力的搜索区域。 在终端窗口中运行以下命令。
pymol AutoDock.py -b receptor.pdbqtIt will take a few minutes for PyMOL to process the imported Python function and load the target protein. Once loaded, you can use the mouse to select the molecule center and then type the following in the PyMOL command line to define your docking analysis search space.
PyMOL需要几分钟来处理导入的Python函数并加载目标蛋白质。 加载后,您可以使用鼠标选择分子中心,然后在PyMOL命令行中键入以下内容以定义对接分析搜索空间。
Box("sele", x, y, z)Where x, y, and z are the magnitudes of the search space in one direction from the center, along the respective axis. The dimensions are in angstroms.
其中,x,y和z是沿着相应轴从中心开始的一个方向上的搜索空间的大小。 尺寸以埃为单位。

Keep trying new search box sizes until you get your desired search area. The smaller the search area, the faster the binding analysis. You will need to delete the box and position entities each time you define a new search space. Make note of the molecule center coordinates and your search box dimensions.
继续尝试新的搜索框大小,直到获得所需的搜索区域。 搜索区域越小,绑定分析越快。 每次定义新的搜索空间时,都需要删除框和位置实体。 记下分子中心坐标和您的搜索框尺寸。
准备配体 (Prepare Ligands)
Our AI generative model output the sampled molecules in the SMILES format. They need to be converted to the .pdbqt file format for our docking analysis.
我们的AI生成模型以SMILES格式输出采样的分子。 需要将它们转换为.pdbqt文件格式,以进行对接分析。
First, convert your SMILES to .sdf files using smi_sdf_ligands.py. This Python program allows the user to provide a ligand family name, increments the name (i.e. name-1, name-2) and assigns it to each .sdf output file. The program also creates a name-to-SMILES mapping spreadsheet so that you can map your docking analysis results back to their original SMILES molecule.
首先,使用smi_sdf_ligands.py将SMILES转换为.sdf文件。 该Python程序允许用户提供一个配体族名称,将该名称(即name-1,name-2)递增,并将其分配给每个.sdf输出文件。 该程序还会创建一个名称到SMILES的映射电子表格,以便您可以将对接分析结果映射回其原始的SMILES分子。
Now convert the .sdf files to .pdbqt files using vina_ligands.py. This program batch coverts the .sdf files to .pdbqt files and adds polar hydrogens to each molecule for docking.
现在,使用vina_ligands.py将.sdf文件转换为.pdbqt文件。 此程序批处理将.sdf文件转换为.pdbqt文件,并向每个分子添加极性氢以进行对接。
基线对接分析 (Baseline Docking Analysis)
A virtual docking analysis was performed between Remdesivir and the COVID-19 main protease to define the baseline binding affinity to beat with our AI generated molecules.
在Remdesivir和COVID-19主蛋白酶之间进行了虚拟对接分析,以定义与我们AI产生的分子搏动的基线结合亲和力。

The most negative number indicates the strongest bond between the ligand and the receptor. Remdesivir’s best binding score was -11.8. Note the modes; AutoDock Vina uses a Monte Carlo simulation to cycle through the various ligand bond configurations (mode) for potential docking against a target.
最负数表示配体和受体之间的最强键。 Remdesivir的最佳结合得分是-11.8。 注意模式; AutoDock Vina使用Monte Carlo模拟在各种配体键配置(模式)之间循环,以潜在地与目标对接。
To perform a batch virtual screen against all of the AI generated ligands, place the ligands (.pdbqt files) in a directory called “Ligands”. Then place this directory, receptor.pdbqt and the AutoDock Vina executable in a common folder. Run the following code.
要针对所有AI生成的配体执行批处理虚拟屏幕,请将配体(.pdbqt文件)放置在名为“配体”的目录中。 然后将此目录,receptor.pdbqt和AutoDock Vina可执行文件放在一个公共文件夹中。 运行以下代码。
for file in ./Ligands/*/*; do tmp=${file%.pdbqt}; name="${tmp##*/}"; ./vina --receptor receptor.pdbqt --ligand "$file" --out $name.out --log $name.log --exhaustiveness 10 --center_x 0 --center_y 0 --center_z 10 --size_x 15 --size_y 15 --size_z 15; awk '/^[-+]+$/{getline;print FILENAME,$0}' $name.log >> temp; done; sort temp -nk 3 > Results; rm temp; mkdir logs; mv *.log *.out logsThis script will take each .pdbqt ligand file and perform an AutoDock Vina docking analysis against the receptor.pdbqt file. After each docking analysis, it will output a log file with the binding results per mode and a .pdbqt file (but with a .out extension). When the virtual screening is complete, the script will collect the results from each analysis log file, sort them and place them in a single results file.
该脚本将获取每个.pdbqt配体文件,并针对receptor.pdbqt文件执行AutoDock Vina对接分析。 每次停靠分析后,它将输出一个日志文件和一个.pdbqt文件(带有.out扩展名),该文件包含每种模式的绑定结果。 虚拟筛选完成后,脚本将从每个分析日志文件中收集结果,对其进行排序并将其放置在单个结果文件中。
You need to input your own receptor center (x, y, z) and search bounding box (size x, size y, size z) values; the above are just placeholders. You also need to set the AutoDock Vina ‘exhaustiveness’ value which determines how many Monte Carlo simulations are attempted during the docking analysis. Here, an exhaustiveness value of 32 was used for the first pass screening of the candidate ligands. After the initial screening, the molecules with the best binding scores (our finalist group) were screened again with an exhaustiveness setting of 256.
您需要输入自己的接收器中心(x,y,z)和搜索边界框(尺寸x,尺寸y,尺寸z)值; 以上只是占位符。 您还需要设置AutoDock Vina的“穷举性”值,该值确定在对接分析期间尝试进行多少次Monte Carlo模拟。 在此,穷举值为32,用于候选配体的第一遍筛选。 初步筛选后,结合性得分最高的分子(我们的决赛入围者)以256的穷举度设置再次筛选。
结果 (Results)
The best binding scores from the virtual docking analysis between the AI generated ligands and the COVID-19 main protease are presented below. While the AM-724 ligand came close to the Remdesivir binding score of -11.8; the rest are notably below the baseline score.
AI产生的配体与COVID-19主要蛋白酶之间的虚拟对接分析得出的最佳结合分数如下所示。 AM-724配体接近Remdesivir结合分数-11.8; 其余的明显低于基线分数。

转移学习 (Transfer Learning)
In AI, the concept of ‘transfer learning’ is to utilize a neural network model that has been successfully trained for one task as your starting point for training a model on a similar, but not necessarily identical, task. Similar to using the learned ability to walk as a precursor to learning to run. Applied here, the base model is incrementally trained on a secondary data set in order to fine tune its molecule creation characteristics.
在AI中,“转移学习”的概念是利用已成功完成一项任务训练的神经网络模型,作为在相似但不一定相同的任务上训练模型的起点。 类似于使用学到的行走能力作为学习跑步的先驱。 在此应用的基础模型在辅助数据集上进行增量训练,以微调其分子创建特性。
The set of AI generated molecules with the best binding scores, our finalist group, is used as input data to further train our neural network model. After the baseline model is incrementally trained for a quarter of the original number of training epochs, the above solution process is repeated:
我们的决赛入围者小组将AI结合得分最高的一组分子用作输入数据,以进一步训练我们的神经网络模型。 在对基线模型进行增量训练达到原始训练时期数的四分之一之后,重复上述解决过程:
- Update the standalone models to use the newly trained weights更新独立模型以使用新训练的权重
- Generate new ligand molecules using those models使用这些模型生成新的配体分子
- Prepare the new ligands for docking analysis; nothing needs to be done to the target protein (COVID-19 main protease)准备用于对接分析的新配体; 不需要对目标蛋白(COVID-19主蛋白酶)进行任何处理
- Perform an initial and final virtual screening of the new molecules对新分子进行初始和最终虚拟筛选
最终结果 (Final Results)
The transfer learning model generated 43 ligands with binding affinity scores better than Remdesivir; the top 10 are denoted below.
转移学习模型产生了43个配体,其结合亲和力得分优于Remdesivir。 前10名如下所示。

We can see that use of transfer learning has improved the neural network model’s ability to generate more desirable ligand inhibitors of the COVID-19 main protease.
我们可以看到,转移学习的使用提高了神经网络模型生成COVID-19主要蛋白酶的更理想配体抑制剂的能力。
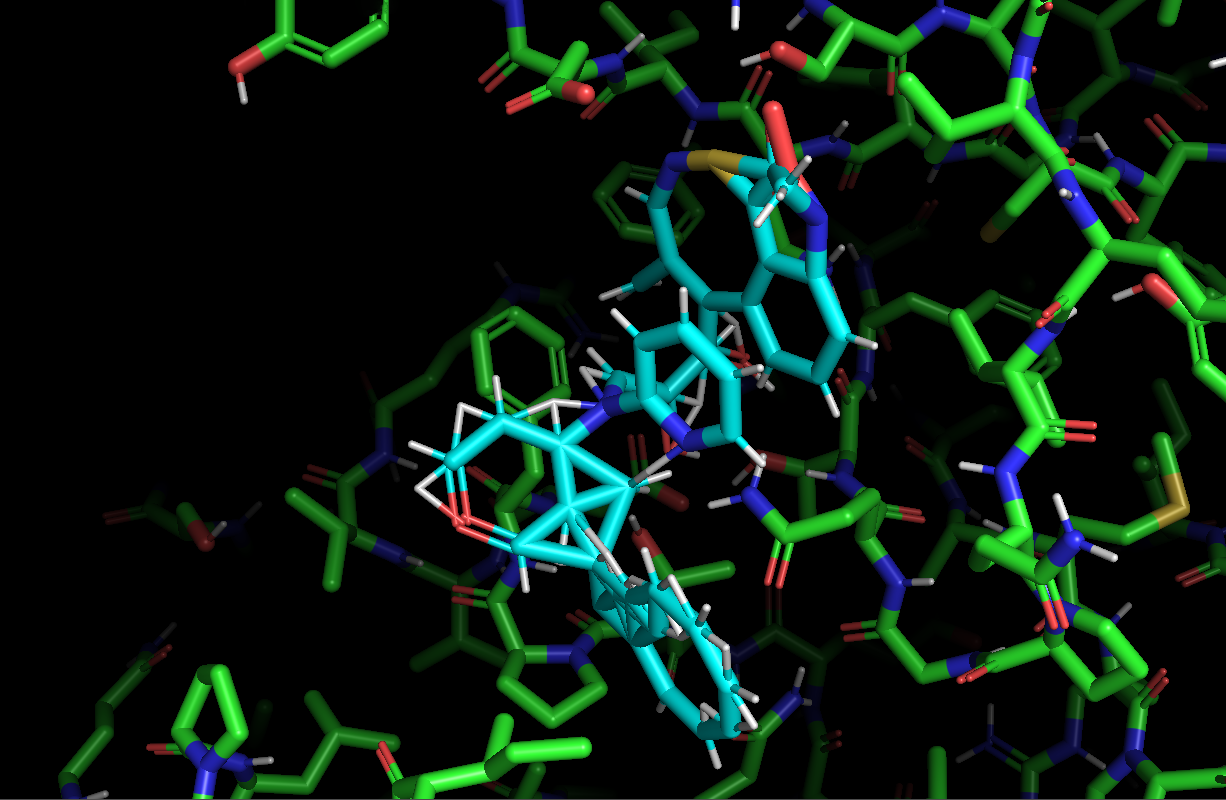
The images below show different views of the ALM-217 ligand docked to the COVID-19 main protease.
下图显示了对接至COVID-19主蛋白酶的ALM-217配体的不同视图。




结论 (Conclusions)
Computational de novo drug design can be useful in expediting either the direct creation of an inhibitor or for providing insights toward existing solutions. Deep learning AI can be used to explore the synthetic molecule solution space and generate novel molecules for target binding.
从头计算药物设计可用于加快抑制剂的直接生产或提供对现有解决方案的见解。 深度学习AI可用于探索合成分子溶液空间并生成用于目标结合的新型分子。
Computational design exists in the realm of virtual space. A domain expert will need to analyze the top findings, assess the feasibility of ligand creation and confirm their performance in a wet lab. Hopefully, this provides some information to kick start that effort. If you are in that field, feel free to reach out to me for the detailed information on the top contenders.
计算设计存在于虚拟空间领域。 领域专家将需要分析主要发现,评估配体创建的可行性并确认其在湿实验室中的性能。 希望这提供了一些信息来启动这项工作。 如果您在该领域,请随时与我联系以获取有关顶级竞争者的详细信息。
下一步 (Next Steps)
The straight forward extension is to repeat the transfer learning/molecule generation / virtual screening cycle through future generations of ligands. Additionally, a systematic exploration of the molecule creation hyperparameters should be performed to improve the binding results.
直截了当的扩展是通过配体的后代重复转移学习/分子生成/虚拟筛选循环。 另外,应该对分子产生超参数进行系统的探索,以改善结合结果。
A more challenging endeavor would be to utilize reinforcement learning / deep Q learning to encapsulate the process whereby the resulting binding affinity score could drive the reinforcement learning policy.
更具挑战性的工作将是利用强化学习/深度Q学习来封装该过程,由此产生的绑定亲和力得分可以驱动强化学习策略。
致谢 (Acknowledgements)
Since no man is an island, some acknowledgments:
由于没有人是一个孤岛,因此要承认以下几点:
- Dr. Andrew Ng (@AndrewYNg) for the excellent AI educationNg博士(@AndrewYNg)出色的AI教育
- Dr. Esben Jannik Bjerrum for inspiration of the deep learning modelsEsben Jannik Bjerrum博士对深度学习模型的启发
- Dr. Sari Sabban for the much needed AutoDock Vina assistanceSari Sabban博士为急需的AutoDock Vina帮助提供了帮助
翻译自: https://medium.com/swlh/ai-de-novo-molecule-design-aed30465e293
de novo转录组 流程
http://www.taodudu.cc/news/show-4762408.html
相关文章:
- 【转】5.3 Python的科学计算包 - Numpy
- TCMalloc 安装和使用
- MAC添加SSH到GitHub
- SCTK的使用——MgB2
- SQLServer日期函数的使用
- 函数内部的this指向/call()方法
- 视频剪辑PR各种版本
- 微信小程序----第四天(基础加强)
- 微信小程序自定义头部导航nav
- 微信小程序流量主提升ecpm的一些方法
- 微信小程序基础库
- matplotlib 绘制并列饼状图
- vue实现3D饼状图
- 用python制作饼状图
- 饼状图环形图数据信息PR图形模板MOGRT
- PB 饼状图制作过程
- python使用matplotlib绘制饼状图
- 高通Ziad Asghar:AI处理的重心从云端向边缘侧转移,智能手机是最佳平台 | MEET 2023...
- 新版gsp计算机系统全套资料,新版gsp全套记录表格120个-20210627023415.pdf-原创力文档...
- 路由器修改dhcp服务器地址,修改路由器dhcp服务器地址
- 华为linux改ip地址,ubuntu9.10校园网上网配置详细教程(华为h3c,mac地址绑定)
- 静态路由知识华为eNSP实践
- 基于微信小程序的宠物寄养平台小程序
- 科一考试要注意什么?
- 干货:大学考试不挂科的小窍门
- 2020科目一考试口诀_科目一考试技巧口诀顺口溜大全,看一遍就能记住,轻松一次过!...
- c语言程序设计第一次月考考试重点,月考小技巧,助你得高分
- 船员英语老师是面试还是计算机答题,船员英语考试、评估技巧
- 临场考试技巧
- 2020科目一考试口诀_2020驾照科目一考试技巧口诀,速记技巧
de novo转录组 流程_AI-De Novo分子设计相关推荐
- 初步了解:使用JavaScript进行表达式(De Do Do Do,De Da Da Da)
by Donavon West 由Donavon West 初步了解:使用JavaScript进行表达式(De Do Do Do,De Da Da Da) (A first look: do expr ...
- Accurate self-correction of errors inlong reads using de Bruijn graphs LoRMA使用de Bruijn图对长read中的错误
Accurate self-correction of errors inlong reads using de Bruijn graphs LoRMA使用de Bruij ...
- 分子排列不同会导致_生物信息遇上Deep learning(7): ReLeaSE--强化学习做药物分子设计...
前言 这篇论文来自北卡罗来纳大学的医学团队的一篇使用强化学习(reinforment learning,RL)技术做药物分子设计的文章,这篇文章和基因组学的关联并不是太大,因为目前在各大期刊上还没找到 ...
- Survey | 基于生成模型的分子设计
今天给大家介绍MIT的Rafeal Gomez-Bombarelli教授发表在arXiv上的综述文章.文章对分子生成模型进行了分类,并介绍了各类模型的发展和性能.最后,作者总结了生成模型作为分子设计前 ...
- Nat. Mach. Intell. | 少量数据的生成式分子设计
今天介绍苏黎世联邦理工大学Gisbert Schneider团队在nature machine intelligence 2020上发表的论文,该论文利用分子语言模型,结合三种优化方法,可以用少量分子 ...
- 驰骋工作流引擎设计系列04 流程引擎表结构的设计
第1节. 关键字 驰骋工作流引擎 流程快速开发平台 workflow ccflow jflow 第1节. 流程引擎表结构的设计 流程引擎表是流程引擎控制流程运转的数据存储表,是整个流程引擎的核心表.理 ...
- 计算机模拟多孔碳,多孔碳材料分子设计的三种方法
原标题:多孔碳材料分子设计的三种方法 多孔固体分子设计在过去二十年里中取得了巨大的成功.得益于其独特的性质,多孔碳材料(尤其是活性碳 )由于其独特的性质已可作为分离介质持续使用.这篇发表于Elsevi ...
- 计算机模拟分子设计,计算机模拟分子材料.pdf
计算机模拟分子材料.pdf 计算机模拟与材料分子设计计算机模拟与材料分子设计 CCCCompuomputttterer SiSiSiSimumullllaatititition anon andddd ...
- 计算机辅助药物设计自学,《计算机辅助药物分子设计》教学大纲
课程名称(中文):计算机辅助药物分子设计 课程名称(英文):Computer-Aided Drug Design 课程编码:211306 开课学院:理学院 授课对象:硕士研究生 任课教师:付颖 学时: ...
最新文章
- DecimalFormat 的使用
- QIIME 2用户文档. 13训练特征分类器Training feature classifiers(2019.7)
- 企业分布式微服务云SpringCloud SpringBoot mybatis (九)Spring Boot多数据源配置与使用(JdbcTemplate支持)...
- LeetCode Interleaving String(动态规划)
- C语言中的数据类型在VB中的申明
- 反射获取有参数的成员方法并运行
- 音频电平vu显示表软件下载_音频控制软件-SoundSource 4 Mac
- 角速度求积分能得到欧拉角吗_高中物理竞赛典型例题精讲——中垂平面内电荷圆周运动角速度...
- 前端面试js-手写事件委托(一点小改进)
- arcgis图层叠加不匹配
- Sql Update Alter Rename
- 深入剖析Redis高可用集群架构原理
- 数据结构练习题――中序遍历二叉树
- 计算机网络wifi是什么意思,wifi的ssid是什么
- 【解析】Token to Token Vision Transformer
- 高考还有几天c语言作业,高考考几天
- Spring实战学习笔记
- DSP TMS320C5509A 控制DDS AD9854芯片进行AM幅度调制
- 全球十大正规外盘期货交易APP平台排名(2022版)
- python实现Hangman游戏