PICRUSt2:OTU或ASV等16S随便预测宏基因组且数据库增加10倍
文章目录
- 简介
- 工作流程
- 引用
- 安装
- 一条命令完成分析
- 核心输出结果:
- 额外输出文件:
- 参数详解
- 猜你喜欢
- 写在后面
PICRUSt推出了近6年,引用2500余次。
现推出PICRUSt2 https://github.com/picrust/picrust2
具有以下三大优势:
- 任何OTU/ASV直接预测功能;
- 数据库扩大10倍;
- 一条命令完成全部分析。
简介
https://github.com/picrust/picrust2/wiki
PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)是一款基于标记基因序列来预测功能丰度的软件。
“功能”通常指的是基因家族,如KEGG同源基因和酶分类号,但可以预测任何一个任意的特性。同样,预测通常基于16S rRNA基因测序数据,但也可以使用其他标记基因。
在这个帮助文档中,您可以找到脚本、安装说明和工作流的描述。有关详细信息,请参见github wiki的右侧栏。
PICRUSt2包括这些改进以及与原始版本相比的其他改进:
- 允许用户预测任何16S序列的功能。来自OTU或扩增序列变体(amplicon sequence variants,ASV,例如DADA2和Deblur输出)的代表性序列可通过序列放置方法用作输入。
- 用于预测的参考基因组数据库扩大了10倍以上。
- 从Castor R包中添加隐藏状态预测算法。
- 允许输出MetaCyc 本体预测,这将可与普通宏基因组学的结果比较。
- 通路丰度的推断现在依赖于MinPath,这使得这些预测更加严格。
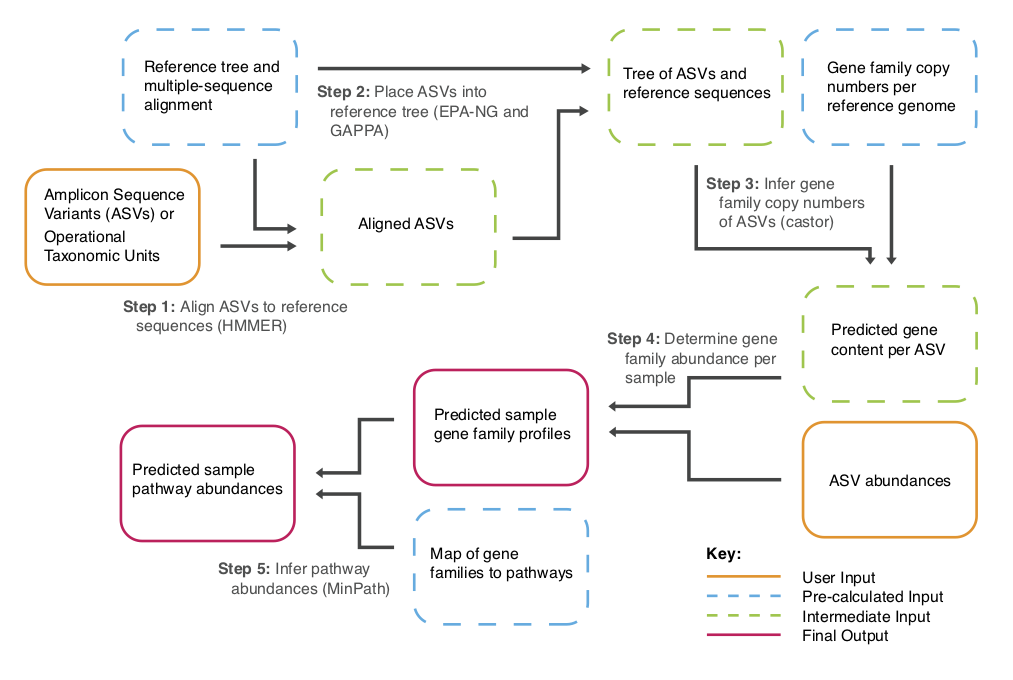
工作流程
PICRUSt2 Flowchart
引用
- For phylogenetic placement of reads:
- HMMER (paper, website)
- EPA-NG (paper, website)
- gappa (pre-print, website).
- For hidden state prediction:
- castor (paper, website)
- For pathway inference:
- MinPath (paper, website)
安装
https://github.com/picrust/picrust2/wiki/Installation
仅支持Linux或Mac,且运行至少16G内存。
推荐conda安装,自动解决依赖关系。
conda create -n picrust2 -c bioconda -c conda-forge picrust2
source activate picrust2
可选源码安装、pip安装,不推荐,详见上方原始网页链接。
wget https://github.com/picrust/picrust2/archive/v2.1.0-b.tar.gz
tar xvzf v2.1.0-b.tar.gz
cd picrust2-2.1.0-b/
conda env create -f picrust2-env.yaml
source activate picrust2
pip install --editable .
测试命令(bioconda安装不可用)
pytest
一条命令完成分析
全部流程已经封装为1个脚本,可以实现上面流程图中的4个主要步骤:
- 进化树中的序列位置确定;
- 预测基因组;
- 预测宏基因组;
- 通路预测;
输出文件为基于16S rRNA基因数据预测的宏基因组。输入文件为fasta文件的代表性序列,可以是OTU或ASV,如下面的study_seqs.fna。还需要一个biom格式或制表符分隔的文本格式的特征表study_seqs.biom。
计算每条序列的最近序列物种索引(NTSI),如果ASV的NTSI > 2将被在分析中排除。--stratified参数将计算层化的输出,但将会极大增加计算时间。
picrust2_pipeline.py -s study_seqs.fna \-i study_seqs.biom \-o picrust2_out_pipeline \--threads 1
比如基于我们常用的代表序列otus.fa和特征表otutab.txt
picrust2_pipeline.py -s otus.fa -i otutab.txt \-o picrust2_out --threads 8
# 1线程42分钟,8线程12分钟,
流程将产生所有结果,包括中间文件(方便用于解决中间出现的问题)将会输出在picrust2_out_pipeline目录中。注意这是默认的输出,你可以指定不同的注释数据库,或自定义的参考数据库(包括非16S数据库)。
核心输出结果:
- EC_metagenome_out - 目录包括非分层的预测宏基因组EC数量 (pred_metagenome_unstrat.tsv), 基于预测16S拷贝数校正的特征表 (seqtab_norm.tsv), 每个样本的NSTI权重 (weighted_nsti.tsv)
- KO_metagenome_out - 和 EC_metagenome_out 类似, 但为宏基因组KO表
- pathways_out - 文件夹包括预测的通路丰度和覆盖度,基于EC数量丰度。
额外输出文件:
可能对进一步分析的经验用户更有用:
- EC_predicted.tsv - 预测的 EC 数量和ASV的拷贝数;
- intermediate - 目录包括MinPath中间文件,用序列取代流程的文件(包括JPLACE文件: intermediate/place_seqs/epa_out/epa_result.jplace).
- KO_predicted.tsv - 和EC_predicted.tsv类似, 为KO 预测中间文件.
- marker_nsti_predicted.tsv - 16S预测的拷贝数和NSTI
- out.tre - 参考序列的树文件,这个树应该比你自己建的树更专业
参数详解
-s PATH - OTU或ASV的序列文件
-i PATH - 序列丰度表 (BIOM, TSV, or mothur shared file format)
-o PATH - 输出目录
–threads INT - 线程数 (默认: 1).
–ref_dir DIRECTORY: 参数用于指定非标准参考序列文件,需要文件夹中包括四个文件如下:
–in_traits IN_TRAITS - 逗号分隔列表(无空格),包括来自以下数据集的基因家族: COG, EC, KO, PFAM, TIGRFAM。注意,这些EC数据默认预测,可用--no_pathways关闭 (默认: EC,KO).
–custom_trait_tables PATH - 可选的目录用于存放性状表,要求包括基因组为行,基因家族为列 (优先级高于 --in_traits setting),用于hidden-state预测。多个表可以用逗号分隔输入,第一个自定义的表用于推断通路丰度。此命令通常还需要一个标记基因表 (–marker_gene_table)
–marker_gene_table PATH - 标记基因拷贝数表 (默认16S 拷贝数).
–pathway_map MAP - 最小通路表 MinPath mapfile. 默认为件为原核生物通路的MetaCyc反应 The default mapfile maps MetaCyc reactions to prokaryotic pathways (default: picrust2/default_files/pathway_mapfiles/metacyc_path2rxn_struc_filt_pro.txt).
–no_pathways - 不进行推断通路,默认为计算的 Flag to indicate that pathways should NOT be inferred (otherwise they will be inferred by default). Predicted E.C. number abundances are used to infer pathways when default reference files are used.
–regroup_map ID_MAP - 基因ID与基因家族对应表,默认的EC编号与MetaCyc反应的对应表 Mapfile of ids to regroup gene families to before running MinPath. The default mapfile is for regrouping E. C. numbers to MetaCyc reactions (default: picrust2/default_files/pathway_mapfiles/ec_level4_to_metacyc_rxn.tsv).
–no_regroup - 不进行基因家族的按反应归类。当你使用自定义数据时,推荐使用此参数 Do not regroup input gene families to reactions as specified in the regrouping mapfile. This option should only be used if you are using custom reference and/or mapping files.
–stratified - 在各层级产生分层的表,即功能对应物种来源,这步需要较多计算时间 Flag to indicate that stratified tables should be generated at all steps (will increase run-time).
-a {hmmalign,papara} - 比对序列至多序列比对的程序,默认为hmmalign。 Which program to use for aligning query sequences to reference MSA prior to EPA-NG step (default: hmmalign).
–max_nsti INT - 序列与参考序列的相似度阈值,大于2认为没有相近基因组将排除在分析之外 Sequences with NSTI values above this value will be excluded (default: 2).
–min_reads INT - 每个ASV的最低丰度,默认为1。即低于此丰度的在层化中被视稀释分类, Minimum number of reads across all samples for each input ASV. ASVs below this cut-off will be counted as part of the “RARE” category in the stratified output (default: 1).
–min_samples INT - ASV在样品中出现的频率,默认1。。即低于此频率的在层化中被视为稀释分类, Minimum number of samples that an ASV needs to be identfied within. ASVs below this cut-off will be counted as part of the “RARE” category in the stratified output (default: 1).
-m {mp,emp_prob,pic,scp,subtree_average} - HSP方法选择,mp预测离散性状使用最大简约法,emp_prob预测离散性状使用经验概率,subtree_average预测连续性状使用子树平均,pic预测连续性状使用进化独立比较,scp预测连续性状使用简约平方变换; HSP method to use. “mp”: predict discrete traits using max parsimony. “emp_prob”: predict discrete traits based on empirical state probabilities across tips. “subtree_average”: predict continuous traits using subtree averaging. “pic”: predict continuous traits with phylogentic independent contrast. “scp”: reconstruct continuous traits using squared-change parsimony (default: mp).
–skip_nsti - 不计算最近序列分类索引NSTI,默认在预测拷贝数表marker_nsti_predicted.tsv中添加列; Do not calculate nearest-sequenced taxon index (NSTI), which is added as a column in the predicted marker-gene copy-number table by default (marker_nsti_predicted.tsv).
–no_gap_fill - 预测前不进行空白填充。默认进行空白填充。 Do not perform gap filling before predicting pathway abundances (Gap filling is on otherwise by default).
–coverage - 计算通路的覆盖度,这计算通路有无的另一种方法。这些值只对实验和高级用户有用。这处与HUMAnN2中计算的方法一致。 Calculate pathway coverages as well as abundances, which are an alternative way to identify which pathway are present. Note that these values are experimental and only useful for advanced users. Coverage is also calculated using the same approach as HUMAnN2.
–skip_minpath - 跳过最小通路计算,默认使用MinPath Do not run MinPath to identify which pathways are present as a first pass (MinPath is run by default).
–per_sequence_contrib - 计算每条序列的贡献,即将计算每个个体的通路,只有当–coverage打开时才计算分层的覆盖度。 Option to specify that stratified abundances should be reported in terms of the contribution by each predicted genome rather than how much each genome is contributing to the overall community abundance. In other words, pathway abundances will be calculated for each individual predicted genome. Stratified coverages will only be reported when this option is used (and --coverage is set).
–verbose - 输出计算过程的代码 If specified, print out wrapped commands to screen.
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA
PICRUSt2:OTU或ASV等16S随便预测宏基因组且数据库增加10倍相关推荐
- PICRUSt2分析实战:16S扩增子OTU或ASV预测宏基因组、新增KEGG层级
PICRUSt2分析实战:16S扩增子OTU或ASV预测宏基因组.新增KEGG层级 更新时间:2021年7月8日 PICRUSt推出了近8年,引用5000余次. 现推出PICRUSt2,202年再次霸 ...
- PICRUSt2分析实战:16S扩增子OTU或ASV预测宏基因组EC、通路、KO(200806更新)
PICRUSt2分析实战:16S扩增子OTU或ASV预测宏基因组 更新时间:2020年8月6日 PICRUSt推出了近7年,引用4000余次. 现推出PICRUSt2,再次霸气发表于顶级期刊Natur ...
- PICRUSt2:OTU/ASV等16S序列随意预测宏基因组,参考数据库增大10倍
PICRUSt推出了近6年,引用2500余次. 现推出PICRUSt2 https://github.com/picrust/picrust2 具有以下三大优势: 任何OTU/ASV直接预测功能: 数 ...
- NBT:PICRUSt2预测宏基因组功能
PICRUSt2预测宏基因组功能 PICRUSt2 for prediction of metagenome functions Nature Biotechnology [IF:31.864] 20 ...
- PICRUSt:16S预测宏基因组-扩增子分析锦上添花
写在前面 16S分析能获得的信息比较有限,一般找到差异OTU,就很难再深入分析了. 如何把差异OTU与细菌自身的基因组功能建立联系呢?很多人在这方面做出了努力. PICRUSt就是让16S扩增子分析锦 ...
- PICRUSt:预测宏基因组功能—16S扩增子分析锦上添花
写在前面 16S分析能获得的信息比较有限,一般找到差异OTU,就很难再深入分析了. 如何把差异OTU与细菌自身的基因组功能建立联系呢?很多人在这方面做出了努力. PICRUSt就是让16S扩增子分析锦 ...
- 16S预测宏基因组最强R包-Tax4Fun
之前在公众号的文章<根据16S预测微生物群落功能最全攻略>阅读人数近3000人,有需求的用户还是非常多的.其中提到了4个软件,之前已经介绍了其中非常有特点的三种,分别为: - PICRUS ...
- RiboFR-Seq:将16S rRNA与宏基因组连接的方法
摘 要 16S rRNA扩增子分析和宏基因组测序是研究微生物群落的两种主要的独立方法.近年来,许多研究将这两种方法结合起来使用,但下游的数据分析是分开进行的,在分类和功能上总是产生不一致或冲突的结果 ...
- 16S+功能预测发Sciences:尸体降解过程中的微生物组
Metcalf, J. L., et al. (2016). "Microbial community assembly and metabolic function during mamm ...
最新文章
- laravel mysql注入_laravel中如何利用反射实现依赖注入
- R语言ggplot2可视化在X轴上可视化时间标签实战:可视化时间标签、对时间标签进行旋转
- 【Python学习系列一】Windows下Python及其IDE(eclipse+pydev)安装
- 02 掌握实现数据导入导出的方法 1214
- 【SpringBoot与SpringCloud的一些根本】
- python 求和并排序_Python堆排序原理与实现方法详解
- java.lang.ClassNotFoundException: net.sf.json.JSONObject
- 去除PreferenceScreen过期提示
- 使用pano2vr生成html5全景页面
- 【工具类】java数字金额转换成大写
- 希科系统(CxServer)经济效益和社会效益分析
- excel图表上x轴y轴怎么设置的教程介绍
- 伊拉克COR证书电子电器
- “智慧新电商”全渠道获客+智能服务接待
- ALSA子系统(十七)------支持Type-C耳机驱动
- 赋值过程中的类型转换,以及截断问题
- JQuery学习22篇(事件委托)
- 虚拟麦克风音频输入_塑电竞强音 为专业而声 全新职业级罗技G PRO X游戏耳机麦克风震撼上市...
- 支付宝支付和微信支付
- 解决java -version 系统找不到文件 C:\ProgramData\Oracle\Java\javapath\java.exe的问题