R 绘制 GWAS 研究的 Manhattan 图
曼哈顿图本质上是一个散点图,用于显示大量非零大范围波动数值,最早应用于全基因组关联分析(GWAS)研究展示高度相关位点。它得名源于样式与曼哈顿天际线相似(如下图)。

近几年,在宏基因组领域,尤其是差异OTU结合分类学结果,采用 Manhattan plot 展示有非常好的效果,倍受推崇。
一
曼哈顿图优点
大数据中,即展示数据全貌,又能快速找到目标基因或 OTU,同时可知目标的具体位置和分类、显著程度等信息。绝对高端大气,而且还有内涵。
二
曼哈顿图解读
数据坐标轴介绍,以下图 GWAS 研究结果为例:
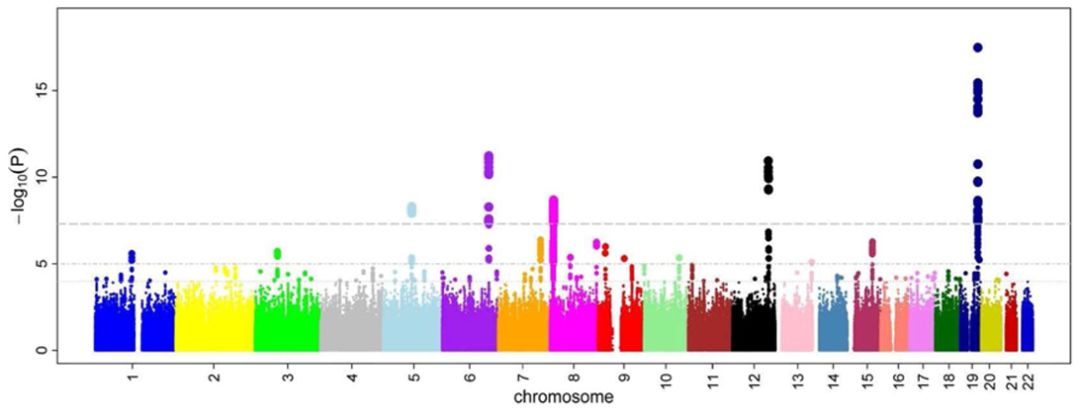
X 轴为染色体编号,且每个基因组 SNP 位点沿染色体序列排列;在 16S 扩增子或宏基因组中则为 OTU 按 Taxonomy 某一级别排序。
Y 轴为该位点相关的统计显著性 Pvalue 值,由于 pvalue 值范围是从 0-1,且越小越好,直接展示非常密集于 0 附近,很难区分。如何使越近 0 的显著数值变大,且而容易区分开,log10 变换是非常好的方法,直接把关注的高显著性(Pvalue 趋近零)值高位显示,远离整体,目标一目了然。
图中水平线一般为设定的不同显著性水平阈值,方便读出每个点的显著性水平;或只添加一条显示性阈值,高于则显著。
三
曼哈顿图绘制工具
R 语言(ggplot2,qqman)、Python(geneview)都能绘制曼哈顿图。本文主要介绍如何使用 R 语言中的 qqman 包绘制 GWAS 研究的 Manhattan 图。
四
R语言 qqman 包
qqman 是一个使用 Q-Q(应用 qq() 函数) 和 manhattan plots(应用 manhattan() 函数) 对 GWAS 分析结果进行可视化的 R 包。
The qqman package includes functions for creating manhattan plots (the manhattan() function) and Q-Q plots (with the qq() function) from GWAS results. The gwasResults data.frame included with the package has simulated results for 16,470 SNPs on 22 chromosomes in a format similar to the output from PLINK.
1. 安装
在 R 中安装 qqman :
# install only onceinstall.packages("qqman")
# load every time you use itlibrary(qqman)【左右滑动查看完整信息】
2. 数据
qqman 包中的 gwasResults 数据结构包含了一共 22 条染色体 16470 个 SNP 的 GWAS 模拟结果数据,该数据共4列,分别是:SNP id,染色体编号,SNP 坐标,P value;如下所示(范例的 SNP 坐标是连续的,但真实的 SNP 数据的坐标可能是断续的,但不会影响绘图):
> str(gwasResults)'data.frame': 16470 obs. of 4 variables: $ SNP: chr "rs1" "rs2" "rs3" "rs4" ... $ CHR: int 1 1 1 1 1 1 1 1 1 1 ... $ BP : int 1 2 3 4 5 6 7 8 9 10 ... $ P : num 0.915 0.937 0.286 0.83 0.642 ...
> head(gwasResults) SNP CHR BP P1 rs1 1 1 0.91480602 rs2 1 2 0.93707543 rs3 1 3 0.28613954 rs4 1 4 0.83044765 rs5 1 5 0.64174556 rs6 1 6 0.5190959
> tail(gwasResults) SNP CHR BP P16465 rs16465 22 530 0.564370216466 rs16466 22 531 0.138286316467 rs16467 22 532 0.393699916468 rs16468 22 533 0.177874916469 rs16469 22 534 0.239302016470 rs16470 22 535 0.2630441【左右滑动查看完整信息】
每一条染色体上的 SNPs 数量:
> as.data.frame(table(gwasResults$CHR)) Var1 Freq1 1 15002 2 11913 3 10404 4 9455 5 8776 6 8257 7 7848 8 7509 9 72110 10 69611 11 67412 12 65513 13 63814 14 62215 15 60816 16 59517 17 58318 18 57219 19 56220 20 55321 21 54422 22 535【左右滑动查看完整信息】
3. 绘制曼哈顿图
绘制最基础曼哈顿图:
manhattan(gwasResults)【左右滑动查看完整信息】
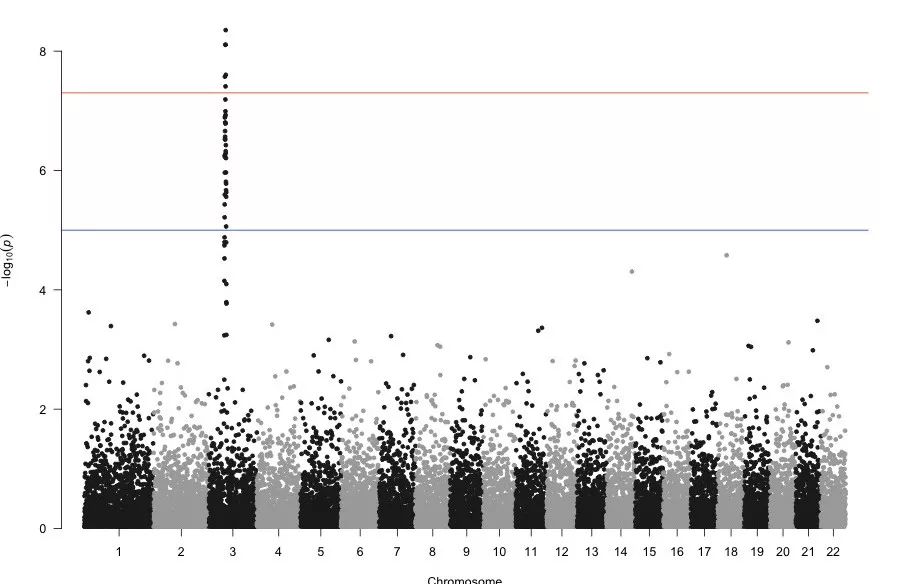
图形说明:
各染色体散点数据来自 gwasResult,是绘图的主数据;
chr3 的线状点来自 qqman 内置的 snpsOfInterest 向量(snpsOfInterest <- paste0("rs", 3001:3100)),由 highlight 参数控制;
灰色横线(suggestive)由参数 suggestiveline 控制,默认 -log10(1e-5) = 5 ;如果为 FALSE,不绘制;
红色横线(genome-wide significance)由参数 genomewideline 控制,默认 -log10(5e-8) ≈ 7.3;如果为 FALSE,不绘制;
灰色、红色横线也可以使用 aline 函数进行绘制:
abline(h=-log10(5e-8), col="red", lty=1, lwd=1)
abline(h=-log10(1e-5), col="blue", lty=1, lwd=1)
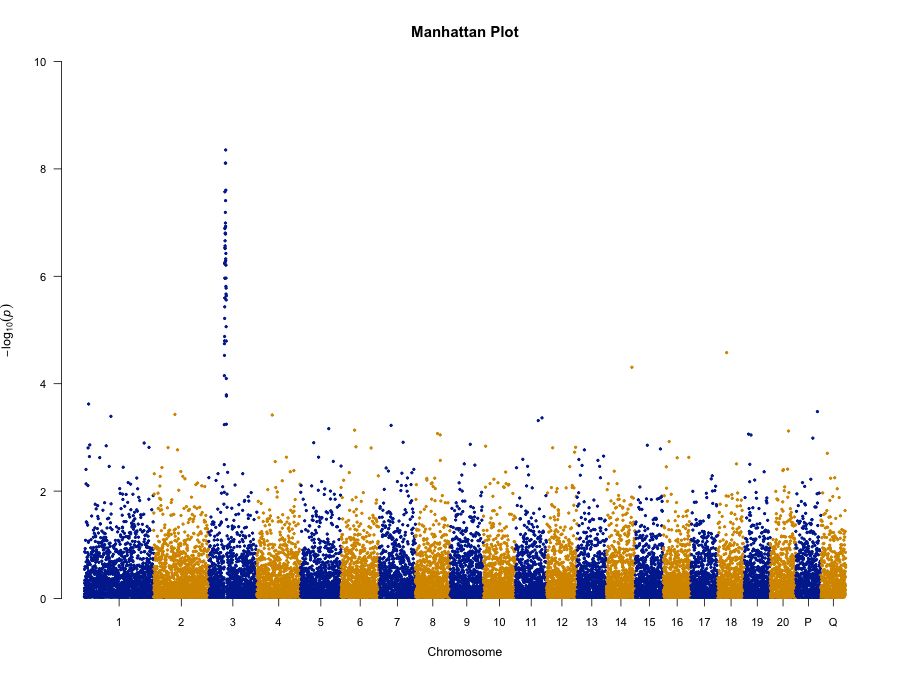
我们可以通过修改参数,更改图形的颜色、标题,以及去除横线:
manhattan(gwasResults, main = "Manhattan Plot", ## 图形标题 ylim = c(0, 10), ## y 轴绘图范围 cex = 0.6, ## 减少点的大小到 60% cex.axis = 0.9, ## 减少 X、Y 轴标签的字体大小到 90% col = c("blue4", "orange3"), ## 散点颜色;该函数会循环利用 col 中的颜色向量 suggestiveline = F, ## 去除 suggestive 参考线 genomewideline = F, ## 去除 genome-wide significance 参考线 chrlabs = c(1:20, "P", "Q")) ## 自定义染色体标号【左右滑动查看完整信息】
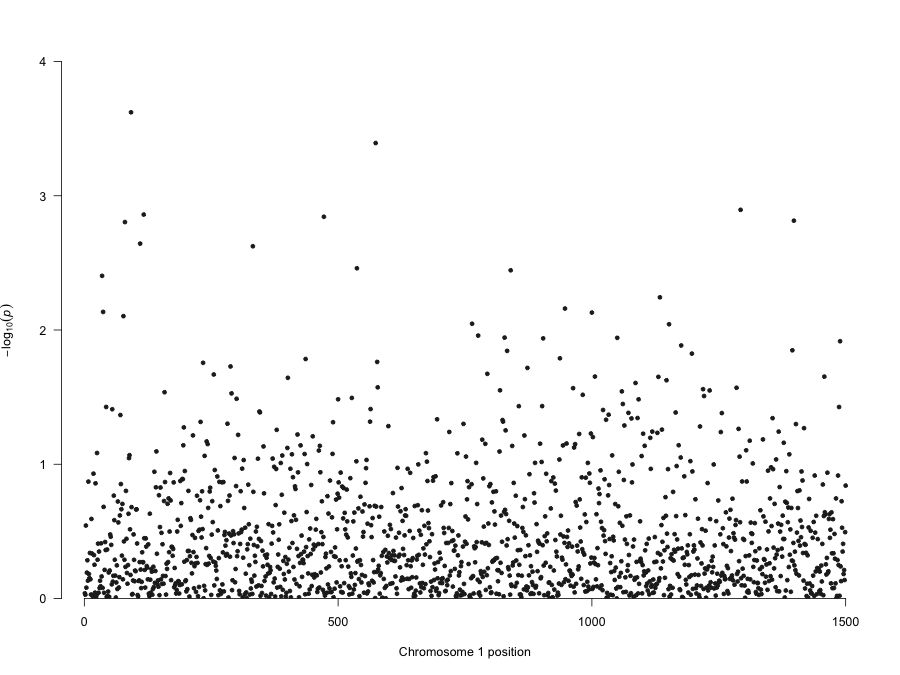
查看单条染色体的分布情况:
manhattan(subset(gwasResults, CHR == 1))【左右滑动查看完整信息】
对 chr3 染色体中感兴趣的 SNPs 进行高亮显示。qqman 包内置了一个叫 snpsOfInterest 向量(vector),该向量包含了 100 个位于 chr3 的 SNPs ( snpsOfInterest <- paste0("rs", 3001:3100) )。
如果我们对不存在的 SNPs 进行高亮(highlight)显示,将会引发警告(warning:You're trying to highlight SNPs that don't exist in your results.)。
> str(snpsOfInterest) chr [1:100] "rs3001" "rs3002" "rs3003" "rs3004" "rs3005" "rs3006" "rs3007" ...> manhattan(gwasResults, highlight = snpsOfInterest)【左右滑动查看完整信息】
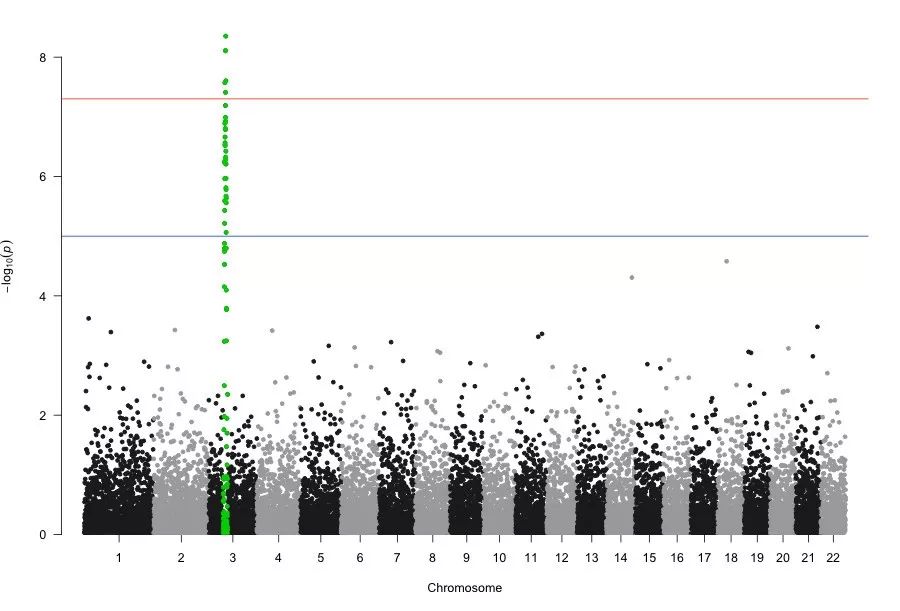
我们也可以自定义感兴趣的 SNPs 进行高亮(highlight)显示,在 GWAS 中如何选择感兴趣的 SNPs:一般是我们做完 gwas 分析,哪个 peak 是你感兴趣的,我们就可以把该 peak 相关的 SNPs 单独提取出来。
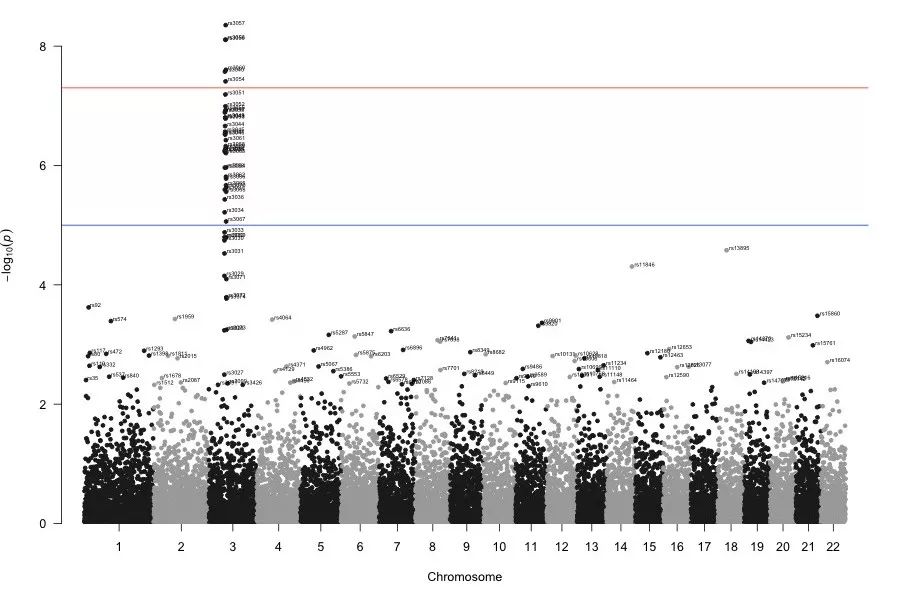
> hig = c("rs3057", "rs3056", 'rs3054', "rs4064", "rs11846", "rs13895")> manhattan(gwasResults, highlight = hig)
#以文件的形式读取感兴趣 SNPs$ cat hig.txtrs3057rs3056rs3054rs4064rs11846rs13895
> hi = read.table("hig.txt")> manhattan(gwasResults, highlight = hi$V1)【左右滑动查看完整信息】
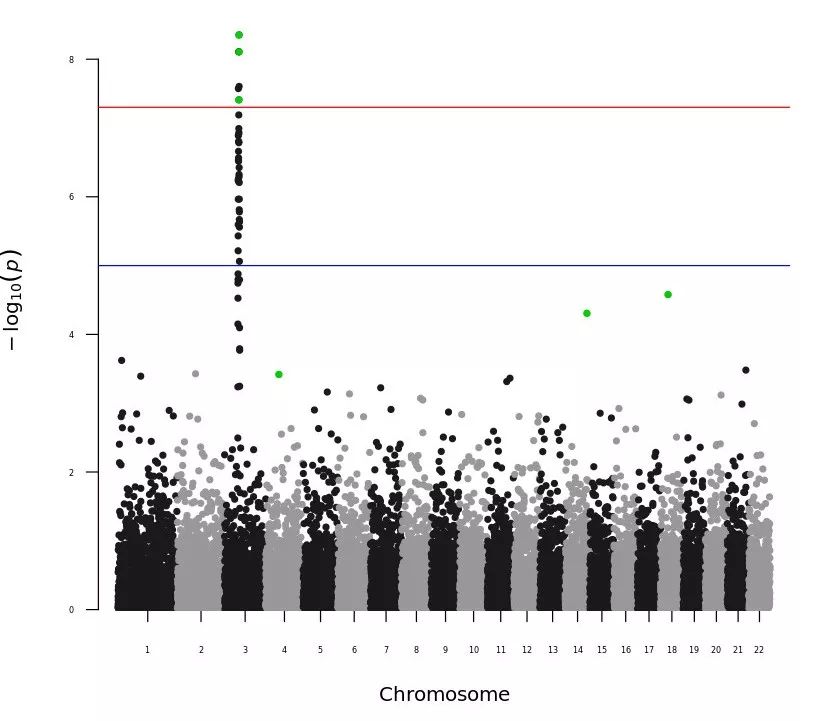
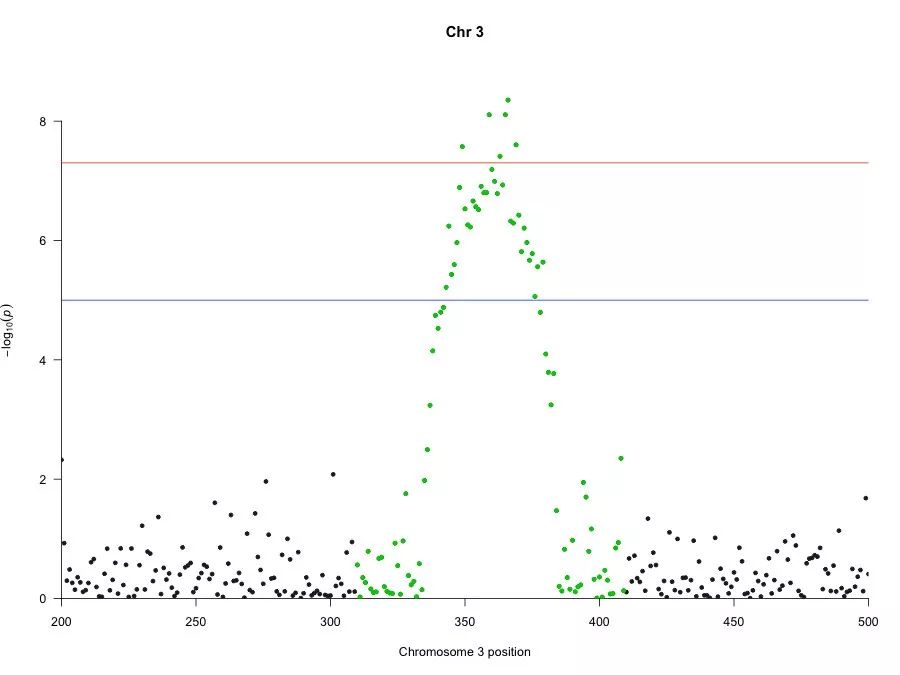
综合高亮和区域限制,我们利用 xlim 参数对位于 chr3:200-500 区域 SNPs 中的 snpsOfInterest 进行高亮显示:
manhattan(subset(gwasResults, CHR == 3), highlight = snpsOfInterest, xlim = c(200, 500), main = "Chr 3")【左右滑动查看完整信息】
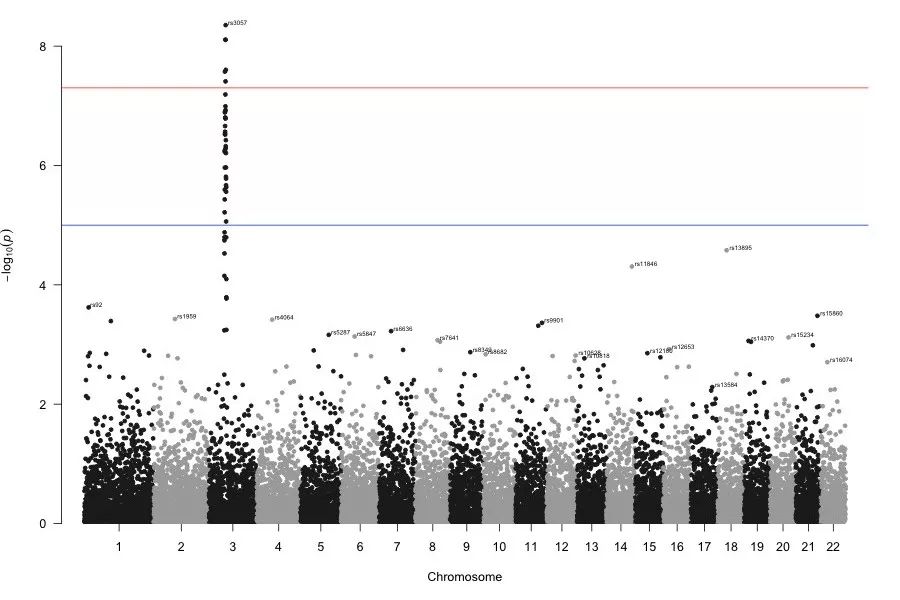
我们可以利用 annotatePval 参数根据 p-value 值对 SNPs 进行注释。该参数默认会对位于每条染色体上,且大于其设定阈值的 Top SNP 进行注释(显示名称):
# -log10(0.001) = 3manhattan(gwasResults, annotatePval = 0.01)【左右滑动查看完整信息】
我们也可以利用 annotatePval+annotateTop 对符合阈值范围内的所有 SNPs 进行注释:
# -log10(0.005) ≈ 2.3manhattan(gwasResults, annotatePval = 0.005, annotateTop = FALSE)【左右滑动查看完整信息】
4. 注意事项
通过执行 str(gwasResults),可以看到 gwasResults 数据框包含了 SNP、染色体、位置,以及 p-value 4 列信息,并分别对应命名为 SNP, CHR, BP,以及 P。如果我们使用 qqman 绘制曼哈顿图但输入数据的列名不一样时,需要使用 chr="输入数据染色体列名", bp="输入数据位置列名",p="输入数据 p-value 列名",以及 snp="输入数据 snp 列名" 进行重新设置。
输入数据的染色体(chromosome)列必须为数值(numeric)。如果名字包含 "X", "Y", 或者 "MT",我们必须把它们重命名为 23, 24, 25 等。我们也可以通过修改源码(e.g., fix(manhattan))的方式更改指定轴刻度标签(labs <- unique(d$CHR))为我们想要的任何形式。
如果想要更改高亮(highlight)或者 suggestive/genomewide 线的颜色,我们需要在源码中找到 col="blue"(suggestive line), col="red"(genomewide line), 以及 col="green3"(highlight colors)后分别进行修改。suggestive/genomewide 线的颜色与样式,也可以使用 aline 函数进行修改(参上上文的"图形说明")。
关于 R 绘制 GWAS 研究的 Manhattan 图就介绍到这里,如果你觉得本文章对你有用,欢迎转发!
本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。
R 绘制 GWAS 研究的 Manhattan 图相关推荐
- R绘制边缘直方图、箱图(Marginal Histogram / Boxplot)
R绘制边缘直方图.箱图(Marginal Histogram / Boxplot) 如果要在同一图表中显示关系和分布(relation and distribution),请使用边缘直方图.它在散点图 ...
- R绘制带显著性标记的热图
今天小编接着来介绍R绘制带有显著性的热图. 测试数据文件: ①table1.txt ②col_group.txt ③row_group.txt 1.导入pheatmap包,读取数据: #导入包 lib ...
- python绘制基因结构图_使用Python绘制GWAS分析中的曼哈顿图和QQ图
[前言]其实这篇文章是为了简单介绍一下geneview的用法,它是一个Python高级库,建立在matplotlib的基础之上,专门用于基因组数据的可视化,目的是为了使创建高大上(精致)的基因组数据图 ...
- r语言把多个图合并在一张图_SAS 绘制亚组分析森林图
背景介绍 随着循证医学(evidence-based medicine)的兴起,森林图(forest plot)已经为人所广泛认识.当前,几乎所有的临床工作者都知道什么是森林图以及如何解读森林图的含义 ...
- stata软件不出图_绘制回归分析结果的森林图,R和Stata软件学起来!
1. 安装forsetplot程序包(绘制森林图)和haven程序包(导入SPSS文件) install.packages("forestplot") install.packag ...
- R语言ggplot2可视化可视化聚类图、使用geom_encircle函数绘制多边形标定属于同一聚类簇的数据点、并自定义每个聚类簇数据点的颜色、多边形框的颜色(Cluster Plot)、主副标题题注
R语言ggplot2可视化可视化聚类图.使用geom_encircle函数绘制多边形标定属于同一聚类簇的数据点.并自定义每个聚类簇数据点的颜色.多边形框的颜色(Cluster Plot).主副标题题注 ...
- R语言ggplot2可视化线图(line plot):当数据有中断、缺失时R不会将数据绘制为连续的线图、而是出现断点
R语言ggplot2可视化线图(line plot):当数据有中断.缺失时R不会将数据绘制为连续的线图.而是出现断点 目录
- R语言使用ggplot2包使用geom_boxplot函数绘制基础分组水平箱图(boxplot)实战
R语言使用ggplot2包使用geom_boxplot函数绘制基础分组水平箱图(boxplot)实战 目录 R语言使用ggplot2包使用geom_boxplot函数绘制基础分组水平箱图(boxplo ...
- R语言ggplot2可视化绘制二维的密度图:在R中建立二维散点数据的连续密度热图、2D密度估计、MASS包中的kde2d函数实现2D密度估计、geom_density2d函数可视化二维密度图
R语言ggplot2可视化绘制二维的密度图:在R中建立二维散点数据的连续密度热图.2D密度估计.MASS包中的kde2d函数实现2D密度估计.geom_density2d函数可视化二维密度图 目录
最新文章
- oracle 学习小结1
- F#创建者Don Syme谈F#设计原则
- centos7安装mysql缺失依赖_CentOS7安装mysql5.7不成功,解决依赖包之后还是无法安装成功...
- 如何为linux 桌面文件内存,linux - 桌面Linux发行版中.desktop文件的功能是什么? - 堆栈内存溢出...
- 【数据结构与算法】之深入解析“把二叉搜索树转换为累加树”和“从二叉搜索树到更大和树”的求解思路与算法示例
- 排序算法 —— 计数排序
- Jackson Annotation Examples
- 78. 子集-LeetCode
- 使用windbg 检查c++程序死锁
- SQL数据库的查询操作大全(select)
- STM32串口通信基本原理
- python 通达信公式函数_通达信zig函数的python实现
- 彻底搞懂ResNet50
- MYSQL报1265和1366错误
- 移动SLAM从入门到入土(二)环境安装
- 《Python编程从入门到实践,P110---习题》
- PMP知识点总结—ADM与PDM的区别
- JASS萌新学习指南1.2
- 芯片破壁者(二):半导体“硅”铺就的文明阶梯
- 润乾报表 动态sql