基因共表达聚类分析及可视化
欢迎关注天下博客:http://blog.genesino.com/2017/11/gene-cluster/
共表达基因的寻找是转录组分析的一个部分,样品多可以使用WGCNA,样品少可直接通过聚类分析如K-means、K-medoids (比K-means更稳定)或Hcluster或设定pearson correlation阈值来选择共表达基因。
下面将实战演示K-means、K-medoids聚类操作和常见问题:如何聚类分析,如何确定合适的cluster数目,如何绘制共表达密度图、线图、热图、网络图等。
获得模拟数据集
MixSim是用来评估聚类算法效率生成模拟数据集的一个R包。
library(MixSim)
# 获得5个中心点,8维属性的数据模型
data = MixSim(MaxOmega=0, K=5, p=8, ecc=0.5, int=c(10, 100))
# 根据模型获得1000次观察的数据集
A <- simdataset(n=1000, Pi=data$Pi, Mu=data$Mu, S=data$S, n.out=0)
data <- A$X
# 数据标准化
data <- t(apply(data, 1, scale))
# 定义行列名字
rownames(data) <- paste("Gene", 1:1000, sep="_")
colnames(data) <- letters[1:8]
head(data)## a b c d e f
## Gene_1 -0.04735251 -0.7147304 0.3836436 1.322786 0.9718988 -0.5468517
## Gene_2 0.09276733 -0.8066507 0.5476909 1.351780 0.8679073 -0.6019107
## Gene_3 -0.08751894 -0.6461075 0.4371506 1.522767 0.8031865 -0.6904081
## Gene_4 0.11065988 -0.7327674 0.4550544 1.379773 0.9304277 -0.5532253
## Gene_5 -0.02722127 -0.7833089 0.6700604 1.448916 0.7128284 -0.6266295
## Gene_6 0.15119823 -0.7468292 0.4859932 1.351159 0.9179421 -0.5625206
## g h
## Gene_1 0.4127370 -1.782130
## Gene_2 0.2852284 -1.736813
## Gene_3 0.3420581 -1.681128
## Gene_4 0.1808146 -1.770737
## Gene_5 0.2936467 -1.688292
## Gene_6 0.1821925 -1.779136K-means
K-means称为K-均值聚类;k-means聚类的基本思想是根据预先设定的分类数目,在样本空间随机选择相应数目的点做为起始聚类中心点;然后将空间中到每个起始中心点距离最近的点作为一个集合,完成第一次聚类;获得第一次聚类集合所有点的平均值做为新的中心点,进行第二次聚类;直到得到的聚类中心点不再变化或达到尝试的上限,则完成了聚类过程。
聚类模拟如下图:

聚类过程需要考虑下面3点:
1.需要确定聚出的类的数目。可通过遍历多个不同的聚类数计算其类内平方和的变化,并绘制线图,一般选择类内平方和降低开始趋于平缓的聚类数作为较优聚类数, 又称elbow算法。下图中拐点很明显,5。
tested_cluster <- 12
wss <- (nrow(data)-1) * sum(apply(data, 2, var))
for (i in 2:tested_cluster) {wss[i] <- kmeans(data, centers=i,iter.max=100, nstart=25)$tot.withinss
}
plot(1:tested_cluster, wss, type="b", xlab="Number of Clusters", ylab="Within groups sum of squares")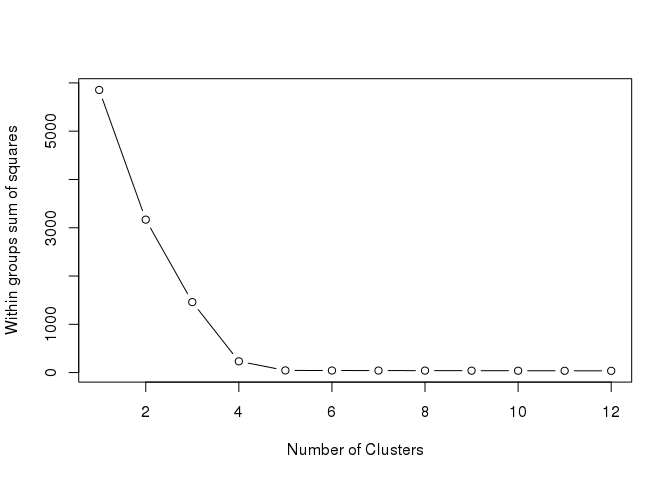
2.K-means聚类起始点为随机选取,容易获得局部最优,需重复计算多次,选择最优结果。
library(cluster)
library(fpc)
# iter.max: 最大迭代次数
# nstart: 选择的随机集的数目
# centers: 上一步推测出的最优类数目
center = 5
fit <- kmeans(data, centers=center, iter.max=100, nstart=25)
withinss <- fit$tot.withinss
print(paste("Get withinss for the first run", withinss))## [1] "Get withinss for the first run 44.381088289378"try_count = 10
for (i in 1:try_count) {tmpfit <- kmeans(data, centers=center, iter.max=100, nstart=25)tmpwithinss <- tmpfit$tot.withinssprint(paste(("The additional "), i, 'run, withinss', tmpwithinss))if (tmpwithinss < withinss){withins <- tmpwithinssfit <- tmpfit}
}## [1] "The additional 1 run, withinss 44.381088289378"
## [1] "The additional 2 run, withinss 44.381088289378"
## [1] "The additional 3 run, withinss 44.381088289378"
## [1] "The additional 4 run, withinss 44.381088289378"
## [1] "The additional 5 run, withinss 44.381088289378"
## [1] "The additional 6 run, withinss 44.381088289378"
## [1] "The additional 7 run, withinss 44.381088289378"
## [1] "The additional 8 run, withinss 44.381088289378"
## [1] "The additional 9 run, withinss 44.381088289378"
## [1] "The additional 10 run, withinss 44.381088289378"fit_cluster = fit$cluster简单绘制下聚类效果
clusplot(data, fit_cluster, shade=T, labels=5, lines=0, color=T,
lty=4, main='K-means clusters')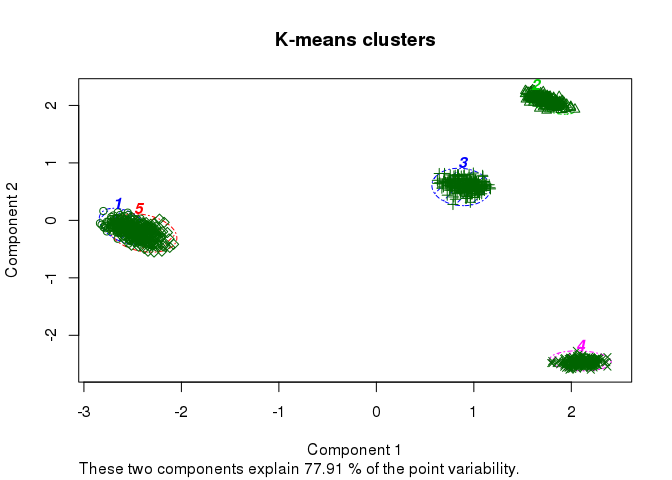
3.预处理:聚类变量值有数量级上的差异时,一般通过标准化处理消除变量的数量级差异。聚类变量之间不应该有较强的线性相关关系。(最开始模拟数据集获取时已考虑)
K-medoids聚类
K-means算法执行过程,首先需要随机选择起始聚类中心点,后续则是根据聚类结点算出平均值作为下次迭代的聚类中心点,迭代过程中计算出的中心点可能在观察数据中,也可能不在。如果选择的中心点是离群点 (outlier)的话,后续的计算就都被带偏了。而K-medoids在迭代过程中选择的中心点是类内观察到的数据中到其它点的距离最小的点,一定在观察点内。两者的差别类似于平均值和中值的差别,中值更为稳健。
围绕中心点划分(Partitioning Around Medoids,PAM)的方法是比较常用的(cluster::pam),与K-means相比,它有几个特征: 1.
接受差异矩阵作为参数;2. 最小化差异度而不是欧氏距离平方和,结果更稳健;3. 引入silhouette plot评估分类结果,并可据此选择最优的分类数目; 4. fpc::pamk函数则可以自动选择最优分类数目,并根据数据集大小选择使用pam还是clara (方法类似于pam,但可以处理更大的数据集) 。
fit_pam <- pamk(data, krange=2:10, critout=T)不同的分类书计算出的silhouette值如下,越趋近于1说明分出的类越好。
## 2 clusters 0.5288058
## 3 clusters 0.6915659
## 4 clusters 0.8415226
## 5 clusters 0.8661989
## 6 clusters 0.7415207
## 7 clusters 0.5862313
## 8 clusters 0.4196284
## 9 clusters 0.2518583
## 10 clusters 0.116984获取分类的数目
fit_pam$nc## [1] 5layout(matrix(c(1, 2), 1, 2))
plot(fit_pam$pamobject)
layout(matrix(1)) #改回每页一张图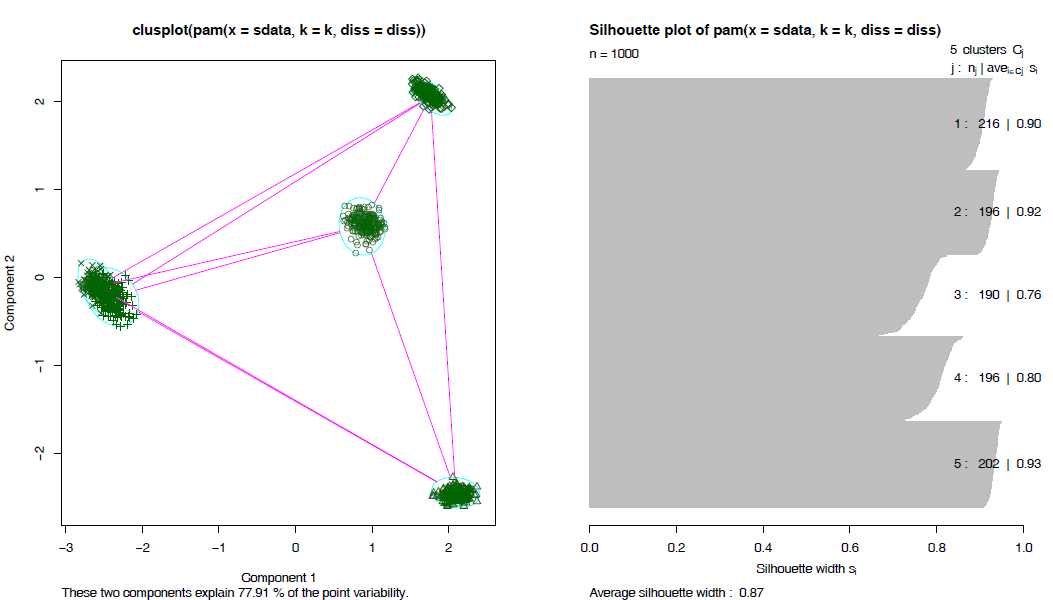
获取分类信息
fit_cluster <- fit_pam$pamobject$clustering数据提取和可视化
以pam的输出结果为例 (上面两种方法的输出结果都已处理为了同一格式,后面的代码通用)。
1.获取每类数值的平均值,利用线图一步画图法获得各个类的趋势特征。
cluster_mean <- aggregate(data, by=list(fit_cluster), FUN=mean)
write.table(t(cluster_mean), file="ehbio.pam.cluster.mean.xls", sep='\t',col.names=F, row.names=T, quote=F)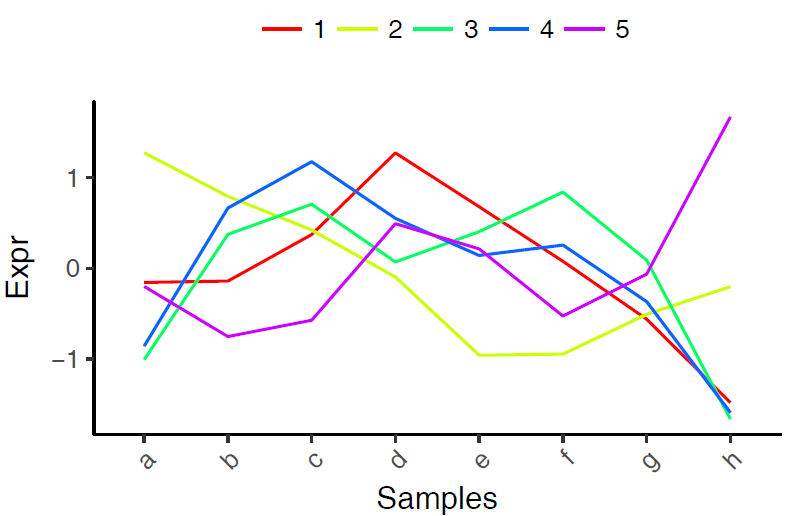
2.获取按照分类排序的数据,绘制热图 (点击查看)。
dataWithClu <- cbind(ID=rownames(data), data, fit_cluster)
dataWithClu <- as.data.frame(dataWithClu)
dataWithClu <- dataWithClu[order(dataWithClu$fit_cluster),]
write.table(dataWithClu, file="ehbio.pam.cluster.xls", sep="\t", row.names=F, col.names=T, quote=F)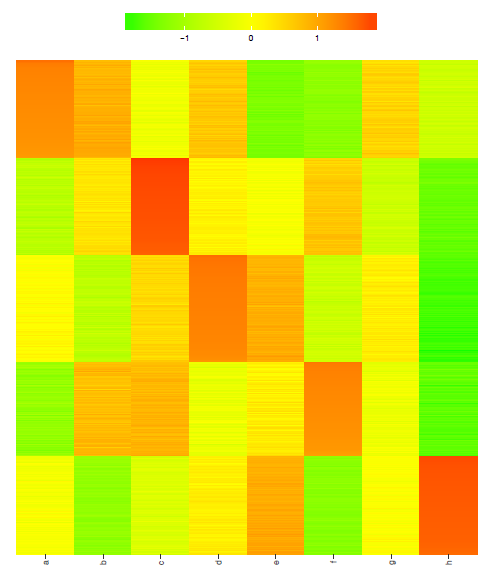
3.获取每类数据,绘制多线图和密度图
cluster3 <- data[fit_cluster==3,]
head(cluster3)## a b c d e f
## Gene_413 -1.2718728 0.6957162 0.9963399 -0.1895966 0.1786798 1.225407
## Gene_414 -1.1705230 0.6765085 0.8689340 -0.2155533 0.4176178 1.251818
## Gene_415 -0.9545339 0.5635188 0.8437158 -0.1360588 0.2084771 1.316728
## Gene_416 -1.0888687 0.8269888 0.7590209 -0.3090701 0.4478664 1.275057
## Gene_417 -1.1230295 0.8282559 0.9112640 -0.2524612 0.3966905 1.104951
## Gene_418 -1.1291253 0.9574904 0.8405449 -0.1200131 0.1964983 1.155913
## g h
## Gene_413 -0.11021165 -1.524462
## Gene_414 -0.22266636 -1.606135
## Gene_415 -0.03562464 -1.806222
## Gene_416 -0.32047268 -1.590522
## Gene_417 -0.21171923 -1.653951
## Gene_418 -0.27792177 -1.623386cluster3 <- t(cluster3)多线图,绘制见线图绘制。
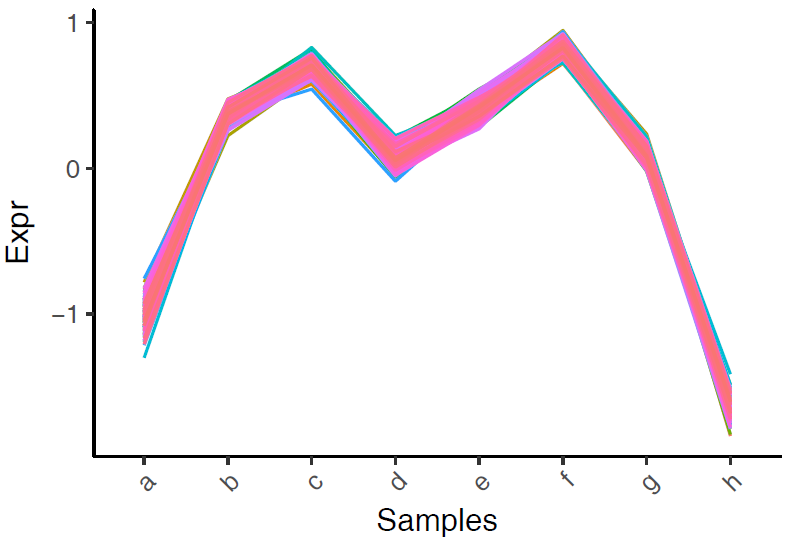
data_rownames <- rownames(cluster3)
data_mean <- data.frame(id=data_rownames, data_mean=rowMeans(cluster3))
data_mean## id data_mean
## a a -1.1708878
## b b 0.7811888
## c c 0.8443212
## d d -0.2008149
## e e 0.2382650
## f f 1.2601860
## g g -0.1612029
## h h -1.5910553library(reshape2)
library(ggplot2)
data_melt <- melt(cluster3)
colnames(data_melt) <- c("id", "Gene", "Expr")
ggplot(data_melt, aes(id, Expr)) + stat_density_2d(aes(alpha=..level.., group=1)) + stat_smooth(data=data_mean, mapping=aes(x=id, y=data_mean, colour="red", group=1), se=F) + theme(legend.position='none')## `geom_smooth()` using method = 'loess'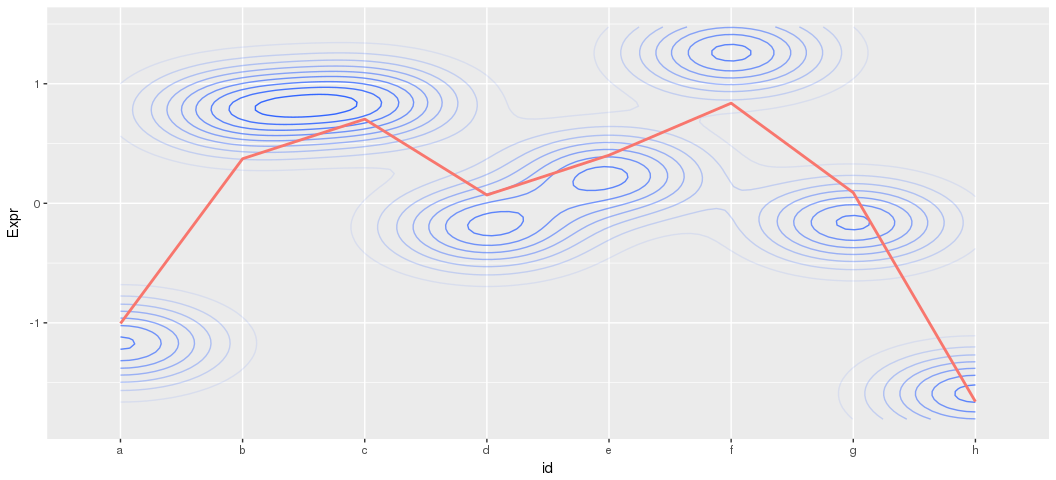
等高线的颜色越深表示对应的Y轴的点越密,对平均值的贡献越大;颜色浅的点表示分布均匀。不代表点的多少。等高线的变化趋势与平均值曲线一致。
参考
- GIF来自 https://commons.wikimedia.org/wiki/File:Kmeans_animation.gif
- PAM更多描述见http://shiyanjun.cn/archives/1398.html
基因共表达聚类分析及可视化相关推荐
- RNA 13. SCI 文章中加权基因共表达网络分析之 WGCNA
WGCNA 分析流程 2008 年发表在 BMC 之后的影响力还是很高的,先后在各大期刊都能看到,但是就其分析的过程来看,还是需要有一定 R 语言的基础才能完整的复现出来文章中的结果,这期就搞出来供大 ...
- Gene co-expression analysis for functional classification and gene–disease predictions 基因共表达分析的功能分类
Gene co-expression analysis for functional classification and gene–disease predictions 基因共表达分析的功能分类和 ...
- 基因共表达网络分析java,基因共表达——基因共表达网络分析
Gene co-expression(基因共表达)是一种使用大量基因表达数据构建基因间的相关性,从而挖掘基因功能的一类分析方法. 在很多情况下,有着相似行为/变化的物质,会存在着一定的联系.在生物中, ...
- WGCNA构建基因共表达网络详细教程
这篇文章更多的是对于混乱的中文资源的梳理,并补充了一些没有提到的重要参数,希望大家不会踩坑. 1. 简介 1.1 背景 WGCNA(weighted gene co-expression networ ...
- 基因共表达网络分析java,WGCNA:加权基因共表达网络分析
加权基因表达网络分析(Weighted gene co-expression network analysis, WGCNA),又叫权重基因共表达网络分析,其根本思想是根据基因表达模式的不同,挖掘出相 ...
- 基因共表达网络分析java,基因共表达网络分析-WGCNA
今天推荐给大家一个R包WGCNA,针对我们的表达谱数据进行分析. 简单介绍:WGCNA首先假定基因网络服从无尺度分布,并定义基因共表达相关矩阵.基因网络形成的邻接函数,然后计算不同节点的相异系数,并据 ...
- WGCNA(加权基因共表达网络分析)
WGCNA(加权基因共表达网络分析) 序章 这个工具现在很火,高分文章用到很多. 加权基因共表达网络分析(WGCNA,Weighted gene co-expression network analy ...
- WGCNA 简明指南|1. 基因共表达网络构建及模块识别
WGCNA 简明指南|1. 基因共表达网络构建及模块识别 参考 简介 数据导入.清洗及预处理 数据导入 检查过度缺失值和离群样本 载入临床特征数据 自动构建网络及识别模块 确定合适的软阈值:网络拓扑分 ...
- WGCNA加权基因共表达网络分析(1)简介、原理
WGCNA简介 WGCNA(Weighted Gene Co-Expression Network Analysis, 加权基因共表达网络分析),鉴定表达模式相似的基因集合(module),解析基因集 ...
最新文章
- Kotlin项目实践指南(上)
- 第十篇 Form表单
- C语言经典例84-一个偶数总能表示为两个素数之和
- 图解Detours实例
- Google MapReduce有啥巧妙优化?
- 目录和文件管理(一)
- 【Python成长之路】来聊聊多线程的几位“辅助”
- 【转】Android虚拟平台的编译和整合
- Mac 版 Android Studio 汉化教程 及汉化包
- golang 编译 执行时候报错cannot declare name db.Query
- Python下载文件到本地
- [转]十年前的老文:以 Linux 的名义
- 基于SpringBoot实现二手交易商城
- FastFDS 分布式文件系统
- codeforces 题解
- C#图片无损转换为ico格式
- w ndows10隐藏桌面设置,据说,这是80%的人都不知道的win10隐藏功能
- linux服务器sftp无法连接超时,sftp连接服务器失败
- 利用python读取tomcat中log文件提取出错误日志生成新的文件
- IEEP-OSPF域内路由故障-现象与排障思路