用k-mer分析进行基因组调查:(一)基本原理
(全文5058字)
【推荐】用Smudgeplot评估物种倍性后,用组合jellyfish+GenomeScope1.0做二倍体物种的基因组调查,用组合KMC+GenomeScope2.0做多倍体物种的基因组调查。
1. 基因组调查(genome survey)
1.1. 基因组调查
基因组调查(genome survey)指基因组特征评估,一般指通过k-mer分析二代测序数据,获得基因组大小(genome size),杂合度(heterozygosity),重复序列比例,GC含量等基因组信息的手段。
基因组调查介绍
在无参考基因组的情况下,使用中低深度的短片段测序所得的reads进行基因组调查(genome survey),初步评估基因组特征,包括基因组大小(genome size),杂合度(heterozygosity),重复序列比例,GC含量等,从而为后续的基因组测序、组装和结构注释方案提供参考依据。
基因组调查的目的
基因组调查主要目的是获取两个方面的信息,一个是基因组的大小,一个是基因组复杂程度。
基因组大小(genome size)
因为测序费用是以测序量为单价计算,所以基因组越大,测序费用越高。
基因组复杂程度
基因组越复杂(杂合度越高,重复序列占比越高),意味着测序难度和组装难度越大。
- 基因组调查实践
- 为了准确估计基因组信息,建议测序深度为50X(即预估基因组大小的50倍),最小也不低于25X。
- 做基因组调查和后续做基因组denovo测序需要使用同一个个体,因为有些物种个体间的基因组特征有较大的差异,且同一个个体做survey的illumina数据可用于组装。
- 预估基因组大小可以通过已有研究粗略判断,包括流式细胞研究,近缘种研究,也推荐植物在C值数据库网站里查询。
- 基因组调查(genome survey)常常使用k-mer分析来实现。
1.2. 基因组复杂程度
基因组复杂程序的判断标准包括:基因组大小,倍性,杂合度,重复序列比例,GC含量等。
一般而言,基因组越大,重复序列比例越高; GC含量异常低或异常高,重复序列比例也会很高;多倍体基因组的杂合度高于二倍体。
判断基因组复杂程度可以参考以下经验性标准:
- 简单基因组: 单倍体;或纯合二倍体;或杂合度低于0.5%, 且重复序列低于50%, 且GC含量在35%-65%的二倍体。
- 复杂基因组: 杂合度在0.5%~1.2%之间,或重复序列高于50%,或GC含量异常(<35%或>65%)的二倍体,或者多倍体。
- 高复杂基因组: 杂合度>1.2%;或重复序列占比大于65%。
2. k-mer分析
k-mer分析可以用在生物信息学许多方面,这篇博客的k-mer分析特指用于基因组调查的k-mer分析方法。
2.1. k-mer相关概念
monomeric unit (mer)
单体单元,单位是nt或者bp。
通常用于双链核酸中的单位,100 mer DNA相当于每一条链有100nt,那么整条链就是100bp。
k-mer概念
在生物信息学中,k-mer是指包含在一段序列中的长度为k的子串。
一段长度为L的核酸序列,以一个碱基为步长滑动,一共可以生成(L-K+1)个k-mers。
另外还可以用这段核酸的反向互补序列再生成一次k-mer。
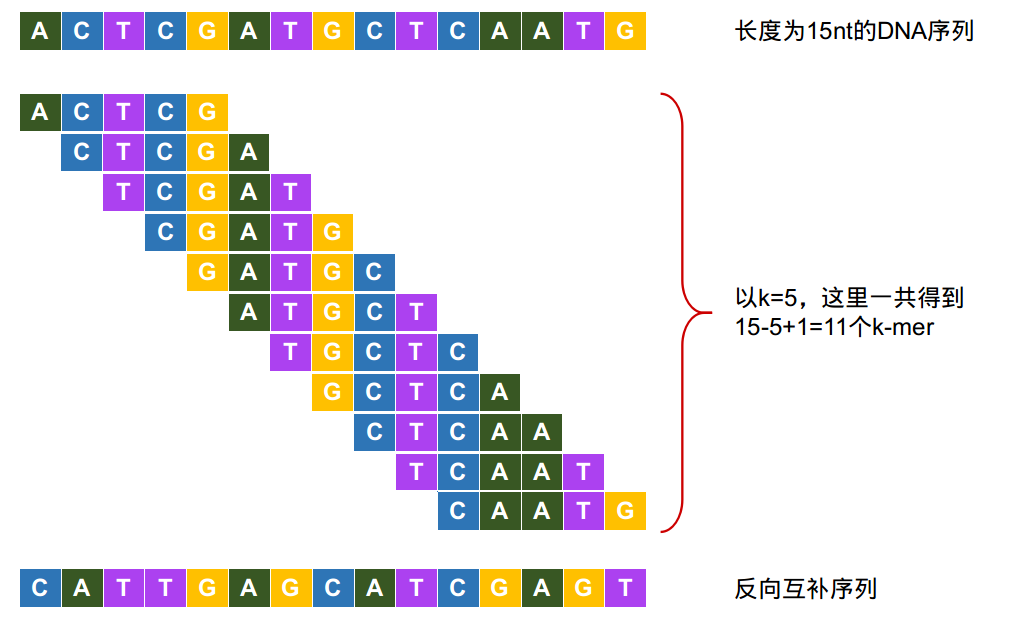
Figure 1. k-mer示例。图片来源:https://cloud.tencent.com/developer/article/1613847
2.2. k-mer分析步骤
- 通过切割二代测序的reads为k-mers
- 统计k-mer的总数和每一种k-mer的频数
- 绘制k-mer的频数分布图
- 根据k-mer的频数分布的主峰峰值判定k-mer的期望深度(即主峰对应的k-mer频数)。
- 根据k-mer的期望深度和k-mer的总数估计基因组大小。
- 根据低频k-mer估计数据错误率,并修正基因组大小的估计。
- 根据k-mer的其他峰估计k-mer的杂合度和重复序列比例。
2.3. k-mer原理
k-mer分析应用的前提假设是测序的reads是随机分布在基因组上的。
首先定义几个变量,方便解释原理:
- 基因组大小:G
- read读长:L
- reads总条数:n
- k-mer长度:K
2.3.1. 碱基深度分布
- 单条read测序覆盖到某一个碱基的概率:L/GL/GL/G。
- 因为L/GL/GL/G很小,n很大,每个碱基覆盖深度服从泊松分布。
- 则每个碱基的覆盖深度的期望为:d=(L/G)∗nd=(L/G)*nd=(L/G)∗n。
2.3.2. k-mer深度分布
一个大小为G的基因组可以产生的k-mer种类约为G。
- 假设一个基因组产生的K都是unique的,从一个大小为G的基因组可以得到(G−K+1)(G-K+1)(G−K+1)种不同的k-mer。
- 一般而言,基因组大小G在几百Mb或者Gb为单位,远大于K和1,所以K和1可以忽略不计,约等于G种k-mer。
单条read测序完全覆盖某种k-mer的概率:(L−K+1)/G(L-K+1)/G(L−K+1)/G。
- k-mer种类的总数约为G
- 单条read能产生的k-mer种类数量为(L−K+1)(L-K+1)(L−K+1)
- 假设read随机分布在基因组上,单条read测序完全覆盖某种k-mer的概率就为:(L−K+1)/G(L-K+1)/G(L−K+1)/G
同样因为(L−K+1)/G(L-K+1)/G(L−K+1)/G很小,n很大,每种k-mer的覆盖深度服从泊松分布。
每种k-mer的覆盖深度的期望为:D=((L−K+1)/G)∗nD=((L-K+1)/G)*nD=((L−K+1)/G)∗n。
由此可以得到,基因组大小为:G=(L−K+1)∗n/DG=(L-K+1)*n/DG=(L−K+1)∗n/D。
2.3.3. 基因组大小
计算总k-mer个数N和k-mer期望深度D。
- 对测序reads进行k-mer分割,获得总k-mer个数N。
- 统计所有分割的k-mer,绘制频数分布图。
- 理想情况下(不考虑测序错误、序列重复性和杂合序列的条件下),k-mer的频数分布遵循泊松分布,可以将频数分布峰值的k-mer频数作为k-mer的期望深度D。
通过下面公式可知,基因组大小G=N/D。
- k-mer的总数量N=(L−K+1)∗nN=(L-K+1)*nN=(L−K+1)∗n
- k-mer的期望深度D=((L−K+1)/G)∗nD=((L-K+1)/G)*nD=((L−K+1)/G)∗n
只要通过k-mer分析计算得到k-mer的总数量N和k-mer的期望深度D,则可以算出基因组大小G。
2.3.4. 基因组调查
在不考虑测序错误、序列重复性和杂合序列的条件下,k-mer的深度分布遵循泊松分布。但实际情况是三者都存在,所以需要计算错误率,重复序列占比和杂合度,并根据计算结果修正对基因组大小的估计。
错误率
- 测序错误:一般认为低频k-mer(K=1,2…)是测序错误引起的,去除低频k-mer并计算错误率以修正基因组大小的估计。
- 一般把拐点前的低频k-mer当作错误去除。
重复序列占比
- 基因组中存在的重复序列会使对应的k-mer频数高于k-mer期望深度D。
- 在k-mer频数分布图中重复序列对应的k-mer会表现为频数大于主峰的一个重复峰。
- 根据主峰右侧的重复峰可以估计基因组的重复序列比例。
- 通过模型计算一个阈值,比如阈值为1.6倍主峰,理论上单拷贝序列(非重复序列)出现在1.6倍主峰右边的概率很低,可以把1.6倍主峰之后的看作重复k-mer。
- 统计重复k-mer的总数量m,从而可以算出:重复序列的大小R=重复k-mer的总数量m/k-mer的期望深度D。
- R/G即为重复序列占基因组的比例。
杂合度
- 基因组中存在的杂合序列会对应两种类型的k-mer,并使得这些k-mer频数低于k-mer期望深度D。
- 假设两种k-mer数量大致相等,在k-mer频数分布图中杂合序列对应的k-mer会表现为频数约为主峰一半的一个杂合峰。
- 根据主峰左侧的杂合峰可以估计基因组的杂合度。
- 假设基因组中的杂合率为 h(每个碱基为杂合点的概率),一个k-mer是纯合k-mer的概率为 P1=(1−h)kP1 = (1-h)^kP1=(1−h)k, 则是杂合的概率 P2=1−P1=1−(1−h)kP2 = 1-P1 = 1-(1-h)^kP2=1−P1=1−(1−h)k。
- 假设产生的k-mer种类的总数为M,其中属于单拷贝序列(非重复序列)的k-mer种类。
- 在二倍体中,单拷贝序列(非重复序列)的同源区域,会产生 U×P2 的杂合k-mer,这个数是可以通过计算得到的:只要统计非重复k-mer的总数 M , M-U即为产生的杂合k-mer数。则 M−U=U×P2=U×(1−(1−h)k)M-U = U×P2 = U×(1-(1-h)^k)M−U=U×P2=U×(1−(1−h)k)。
- 通过计算,即可通过M和U的值得到基因组的杂合率h。
在实际应用过程中,估计了基因组的错误率、杂合度和重复序列比例后,重新修正基因组大小的估计,从而得到基因组调查的结果。

Figure 2. k-mer分析(软件GenomeScope)结果示例
2.4. k-mer用途
许多分析都会用到k-mer的处理方法,把测序得到的reads通过截取k-mer后用于分析。
比如评估基因组特征,组装基因组,物种样品污染评估等。评估基因组特征(genome survey)包括评估基因组大小(size),杂合度,重复序列比例等。
组装基因组
组装基因组使用k-mer的目的主要是去除低频率的k-mer以提高组装结果准确性。
评估基因组大小(size)
在不考虑测序错误、序列重复性的条件下,k-mer的深度分布遵循泊松分布,可以将深度分布曲线的峰值作为期望测序深度。
在k-mer原理部分解释了用k-mer评估基因组大小的公式:
基因组大小G=总k−mer数量N/k−mer期望深度D基因组大小G=总k-mer数量N/k-mer期望深度D基因组大小G=总k−mer数量N/k−mer期望深度D
评估基因组杂合度
根据k-mer频数分布图的杂合峰,可以估计基因组的杂合度。评估基因组重复序列比例
根据k-mer频数分布图的重复峰,可以估计基因组的重复序列比例。
评估物种样品污染
根据k-mer频数分布图,可以评估测序样品污染情况。
- 如果k-mer频数分布图出现两个明显的峰,但两个峰的横坐标又不是二倍关系,那就可能是DNA污染导致的。
- 因为一个物种的杂合峰的频数约为主峰的一半,重复峰的频数约为主峰的两倍。
- 如果DNA中存在两个物种的样品(即有污染),期望会出现对应的两个峰,且两个峰的横坐标之间也非两倍的关系,由此可以初步判定该测序样本存在污染。
- 至于具体是因为什么物种造成污染,则可以通过blast比对nr库进行简单的判断。
除了通过k-mer频数分布评估DNA样品的污染程度外,还可以通过GC含量分布图判断,查看图中是否存在多个密度集中的类群。
2.5. k-mer的优势和局限
增加准确率
- 二代测序的准确率已达到99.9%,但测序量非常大时,错误碱基的绝对数量(比如10亿碱基里错误碱基数量会达到1000万个)还是会对分析有很大的影响。
- 由于测序错误具有随机性,通过将reads切割产生的k-mer中,测序错误生成的k-mer绝大多数都是测序物种中不存在的k-mer,因此都只出现1次(或很少的几次),要是将这些低频的k-mer去掉,就有较大可能去除测序错误,从而使得分析(基因组调查,组装基因组)结果更可靠。
不适用过于复杂的基因组
- k-mer分析适用于分析唯一主峰区域所占比例较大的简单基因组。
- 当基因组杂合非常高或者重复序列比例非常大时,其影响可能导致无法通过k-mer分析正确估计基因组大小。
2.6. k-mer的选择
2.6.1 k-mer的大小选择
- K应该足够大到k-mer可以映射到基因组的唯一位置。
- 太大的k-mer会降低去除低频k-mer代表的错误碱基的概率(增加错误率),也会降低k-mer深度(使得k-mer频数分布的峰不明显),而且大的k-mer会增加计算资源的使用。
- 基因组调查一般选17,21比较常见。
2.6.2 选择k-mer的考虑
- k-mer只能是奇数?
- 把k-mer设置成奇数是为了防止通过k-mer组装时,正反链混淆。
- 偶数的k-mer的反向互补序列常常与自身一样,从而组装k-mer时会混淆正反链的组装,奇数的k-mer就不存在这个问题。
- k-mer的长度代表了可能存在的k-mer种类的数量(4的K次方),越长的k-mer片段映射的物种特异性越强。基因组越大,需要的k-mer越长。
- 当基因组中有较多重复序列时,可以用较大的k-mer来跨过高重复的区域,从而获得更加准确及完整的基因组草图;
- 由于reads上的碱基错误率的存在,选择较长的k-mer会带来较高的错误率,这也可以加大测序深度来弥补。
- 如果是用于组装基因组,为了得到更加完整的基因组,要尽可能使用较长的k-mer用于组装。
3. k-mer分析软件
3.1. k-mer分析软件简介
k-mer分析分为k-mer频数统计和基因组特征评估两步。此外,Smudgeplot还可以用k-mer分析评估物种的倍性。
- jellyfish
- jellyfish可以实现第一步k-mer频数统计。
- 特点是使用Hash表存储数据,能多线程运行;速度快,内存消耗小。
- KMC
- KMC可以实现第一步k-mer频数统计。
- GenomeScope
- GenomeScope可以实现第二步,利用k-mer频数统计结果进行基因组特征评估。
- 1.0版本用于二倍体物种,2.0版本用于多倍体物种。
- KAT(The k-mer Analysis Toolkit)
- KAT(The k-mer Analysis Toolkit)可以实现k-mer频数统计和基因组特征评估两步。
- 包含多个工具来帮助用户通过使用k-mer对测序数据进行简单分析,如组装完整性、测序错误、是否有污染等。
- GCE
- GCE可以分别实现k-mer频数统计和基因组特征评估两步。
- KmerGenie
- KmerGenie可以同时实现k-mer频数统计和基因组特征评估两步。
- 最大优点在于可以实现在多个预设k-mer下的自动分析,除了进行常规的k-mer频数统计之外,还能够基于不同k-mer自动计算基因组大小,并为基因组组装评估一个最佳组装k-mer数值作为备选。
3.2. 一些使用软件的经验总结
- 【推荐】用Smudgeplot评估物种倍性后,用组合jellyfish+GenomeScope1.0做二倍体物种的基因组调查,用组合KMC+GenomeScope2.0做多倍体物种的基因组调查。
- k-mer长度常用17/21。
- 软件KmerGenie,GCE和jellyfish获取的频数分布表,都可用于软件genomescope和GCE第二步骤的分析。
- 由于老版本的GCE第一步骤支持的最大k-mer频数为255,大于255的数据被合并(新版本GCE好像更新了,目前没有这个限制了);而jellyfish统计到10000行,预估结果会更为准确。
- GenomeScope对于高重复序列的基因组统计的基因组大小会偏小,建议max kmer coverage设置大一点,大于等于10000。
- 有些软件(比如GCE)有另一个参数需注意和设定,单倍体模式还是杂合模式,可以两种模式都分析,查看差别。
- 实践经验发现,k-mer值设置得越高,估计出来的基因组size会越大;
- 另外,在jellyfish里的jellyfish histo统计频数分布时,用参数-h 10000把统计上限调高,以及在GenomeScope阶段,Max kmer coverage设置的大一些(即统计进的kmer数量越多),估算出来的基因组大小也会略大一些。
4. references
- wiki_k-mer: https://en.wikipedia.org/wiki/k-mer
- genome survey:https://xuzhougeng.top/archives/genome-survey-using-kmers
- xuzhougeng’s blog:https://www.jianshu.com/p/85de8f025899
- k-mer与基因组组装:https://cloud.tencent.com/developer/article/1613847
- k-mer分析和原理:https://www.bbsmax.com/A/lk5aQMxP51/
- jellyfish paper:https://academic.oup.com/bioinformatics/article/27/6/764/234905?login=true
- jellyfish github:https://github.com/gmarcais/Jellyfish
- GenomeScope1.0 github:https://github.com/schatzlab/genomescope
- KAT github:https://github.com/TGAC/KAT
- GCE github:https://github.com/fanagislab/GCE
用k-mer分析进行基因组调查:(一)基本原理相关推荐
- 生信步骤|kmc+genomescope进行基因组调查
在组装未知基因组时,往往需要利用重测序数据提前进行基因组调查,以获取其基因组规模,杂合率,重复序列比例,GC含量等信息.从而更好地拟定后继测序策略.基因组调查可以采用kmers方法.kmers基因组调 ...
- 分析全基因组上的蛋白信息
背景 在ncbi上查一个细菌的全基因组,搜取全基因组上的这五种信息:gene,locus_tag,note,location,product,蛋白序列的前10个氨基酸(为什么爬这个最后讲),并分析这个 ...
- iMeta视频教程 | StrainPanDA分析宏基因组共存菌株的组成和基因成分谱
点击蓝字 关注我们 StrainPanDA宏基因组分析软件 超详细教程 StrainPanDA主要针对宏基因组纵向数据,能同时实现共存菌株的组成和基因成分谱的获取,方便用户快速建立功能和组成之间的关联 ...
- Week2 Teamework from Z.XML - 必应缤纷桌面助手 - 软件分析与用户需求调查
软件分析与用户需求调查(2013) from Z.XML 本次团队作业要求: 通过定性, 定量地分析, 总结和评定某软件是否满足了目标用户的需求,并把分析的过程和结果用博客表达出来. 选题:必应缤纷桌 ...
- Bioinformatics BIB|港城大孙燕妮组用于识别和分析宏基因组数据中噬菌体序列的网站...
PhaBOX: 用于识别和分析宏基因组数据中噬菌体序列的网站 PhaBOX: a server for identifying and characterizing phage contigs in ...
- Microbiome:NGLess语言实现快速可重复分析宏基因组的流程NG-meta-profiler
文章目录 NG-meta-profiler:使用NGLess(一种领域专用语言)快速处理宏基因组 Peer Bork的介绍和相关文章 日报 摘要 背景 结果 结论 主要结果 图1. NG-meta-p ...
- Week2 Teamework from Z.XML 软件分析与用户需求调查(四)Bing桌面及助手的现状与发展...
一.Bing搜索的相关背景 第一,必应搜索前几年的发展重点在于欧美市场,并且取得了一定的成效:根据 Hitwise 的统计数据,Bing 在 2011年3 月份市场占有率突破了 30% 大关,达到 3 ...
- 必应输入法的分析与用户需求调查报告
0.引言 为响应微软团队提供的用户体验调查,我们远航1617团队分成三个小组,分别对功能.体验.自选这三部分内容对微软必应输入法进行了调查统计与分析建议. 我们团队自认为此次调查的人群比较有代表性,包 ...
- k线分析中的量化测试方法_k线分析中如何使用量化思维案例分析
<量化分析海龟训练营>课程学员:进行程序化交易的前提是什么呢? 同济桥博士:能够进行程序化交易的前提,是能够写成代码进行测试.如果要测试,就要交给电脑去做测试.那么,第一个先决条件,就是要 ...
- 三天实现独立分析宏基因组数据(有参、无参和分箱等)
在广大粉丝的期待下,<生信宝典>联合<宏基因组>在2019年11月1-3日,北京鼓楼推出<宏基因组分析>专题培训第六期,为大家提供一条走进生信大门的捷径.为同行提供 ...
最新文章
- 01-mybatis源码阅读
- 揭秘互联网人群层级,你属于第几级?
- ROS中使用Eigen库[不定期更新]
- JVM01---简介
- jquery获取iframe里的js事件
- 【自用】docker命令记录
- Hi3515主板UBoot参数备份
- opencore(OC引导)关于修改sle系统权限的问题
- orcadcapture安装_电路原理图设计软件(OrCAD Capture CIS 中文版) 16.6 免费安装版
- Hadoop版本区别Hadoop组成
- html表格左边锁定,表头锁定
- 韩泰机器人_韩泰轮胎怎么样?进击的韩泰,拥有着对未来无限的可能!
- Widget我姗姗来迟
- 学习FPGA的网站推荐
- 常用的php函数与类
- 如何在万能地图下载器中购买流量下载地图
- 网络安全实训记录(2020.7.15-2020.7.19)
- db2编目数据库 linux,[转]Db2 catalog(编目)
- JavaScript读写Cookie
- Vue3+TypeScript项目构建之实现自定义指令v-loading, axios请求时的加载动画